8.1. El lisosoma: características generales y morfología
El lisosoma fue descubierto por Christian de Duve en los años 50, quien observó que las células contenían gran cantidad de enzimas hidrolíticos capaces de degradar prácticamente cualquier macromolécula. Dedujo que debían estar compartimentalizados en vesículas membranosas para no destruir la propia célula. En 1974 recibió el Nobel de Fisiología por este descubrimiento.
Los lisosomas son vesículas membranosas de 0,2 a 0,5 μm de diámetro. Su forma y tamaño son variables: es imposible identificarlos al microscopio electrónico solo por su morfología, pues varían mucho entre tipos celulares y según su estado funcional. La única forma de identificarlos con certeza es mediante técnicas citoquímicas que detectan sus enzimas hidrolíticas ácidas características.
8.1.1. Composición y membrana lisosomal
El lumen del lisosoma contiene más de 50 hidrolasas ácidas (proteasas, lipasas, nucleasas, glucosidasas, sulfatasas) capaces de degradar proteínas, lípidos, ácidos nucleicos, polisacáridos y prácticamente cualquier macromolécula biológica. Todas estas enzimas tienen su pH óptimo de actividad en torno a pH 5, muy por debajo del pH citosólico (~7,2). Esto es una medida de seguridad: si los enzimas escapan al citoplasma, el pH neutro los inactiva.
El pH ácido del lumen se mantiene gracias a bombas de protones (V-ATPasas) en la membrana lisosomal que bombean H⁺ al interior de forma activa con gasto de ATP.
La membrana lisosomal tiene propiedades especiales que la protegen de sus propias enzimas:
Está muy glicosilada en su cara luminal: la densa capa de azúcares actúa como escudo protector frente a las proteasas del lumen.
Tiene abundantes proteínas transportadoras que exportan al citoplasma los productos de la digestión: aminoácidos, monosacáridos, nucleósidos, ácidos grasos.
8.2. Funciones del lisosoma: las tres rutas de digestión
El lisosoma recibe material para degradar por tres vías:
8.2.1. Endocitosis
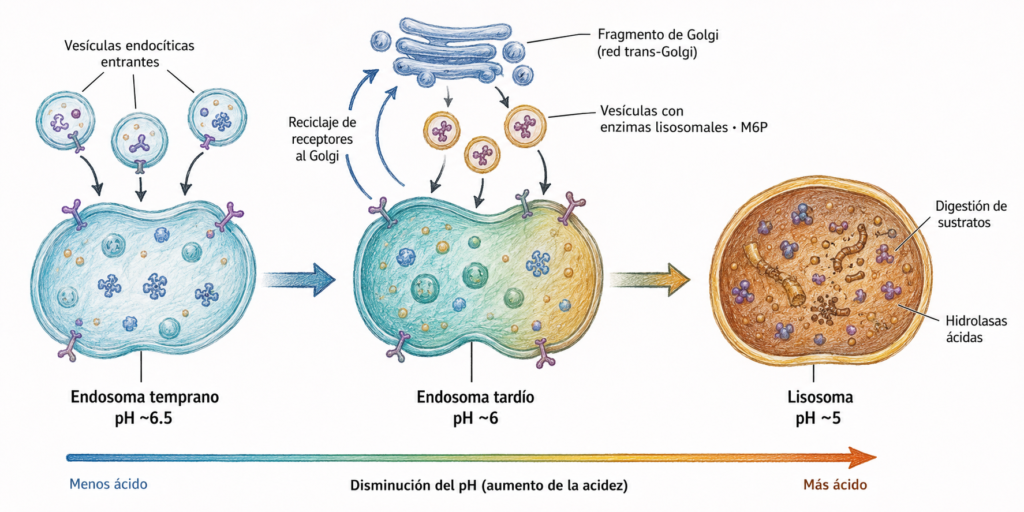
El material endocitado del exterior celular llega al lisosoma a través de la vía endosomal. Las vesículas de endocitosis se fusionan formando el endosoma temprano (pH ~6,5), que madura progresivamente acidificando su lumen hasta convertirse en endosoma tardío (pH ~6). El endosoma tardío recibe vesículas procedentes del aparato de Golgi cargadas de enzimas hidrolíticos y completa su maduración hasta convertirse en lisosoma (pH ~5).
Tras la digestión, los productos útiles se exportan al citoplasma por las proteínas transportadoras de la membrana lisosomal. Los productos no digeribles quedan acumulados en el cuerpo residual. En células especializadas (como los melanocitos), el cuerpo residual puede exocitarse al exterior.
8.2.2. Autofagia
La autofagia es el mecanismo por el que la célula degrada sus propios componentes obsoletos o dañados: orgánulos envejecidos, proteínas agregadas o fragmentos citoplasmáticos. Es esencial para el mantenimiento celular y se activa especialmente en situaciones de estrés, ayuno o infección.
El proceso comienza con la formación de una membrana de doble capa que rodea al orgánulo o componente destinado a la degradación, formando el autofagosoma. El autofagosoma se fusiona con el endosoma tardío o directamente con el lisosoma, donde su contenido es degradado por las hidrolasas.
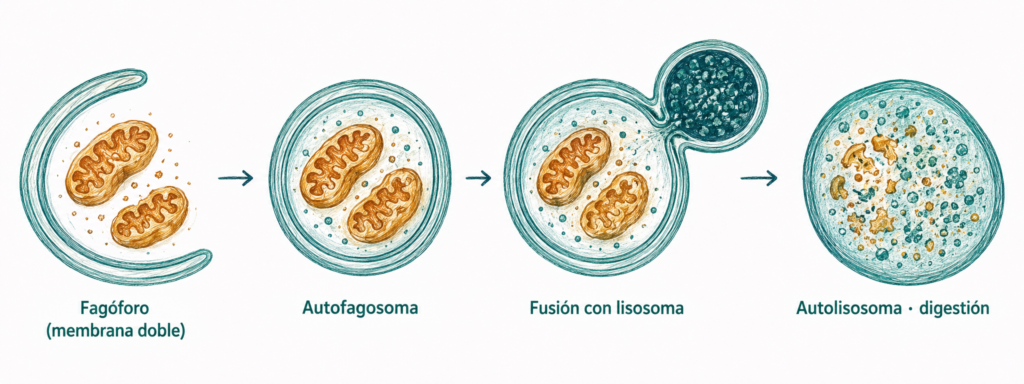
8.2.3. Fagocitosis
La fagocitosis es exclusiva de células especializadas del sistema inmune: macrófagos, neutrófilos y células dendríticas. Estas células engullen bacterias, virus, células muertas y otros materiales extraños formando un fagosoma. El fagosoma se fusiona con el endosoma tardío o con el lisosoma, donde el material es degradado enzimáticamente.
8.3. Biogénesis del lisosoma
Los lisosomas se forman a partir del endosoma tardío tras recibir las vesículas con enzimas hidrolíticos procedentes del aparato de Golgi.
El mecanismo de dirección de las enzimas lisosomales al lisosoma depende del marcador manosa-6-fosfato (M6P):
En la red cis-Golgi, la enzima N-acetilglucosamina fosfotransferasa reconoce una secuencia específica de aminoácidos en las hidrolasas lisosomales y añade una N-acetilglucosamina fosforilada a los residuos de manosa del oligosacárido. Después, la fosfoglucosidasa retira la N-acetilglucosamina, dejando el fosfato unido directamente a la manosa: el marcador M6P.
En la red trans-Golgi, el marcador M6P es reconocido por receptores de M6P en la membrana. Estos receptores concentran las enzimas lisosomales en vesículas recubiertas de clatrina que brotan de la red trans-Golgi. La vesícula pierde la clatrina y se fusiona con el endosoma tardío. La acidificación del endosoma (pH ~6) provoca la disociación de la enzima y su receptor. El receptor es reciclado de vuelta a la red trans-Golgi en vesículas desnudas para ser reutilizado. El fosfato se elimina rápidamente de la manosa, impidiendo el retorno de la enzima al Golgi y activando la hidrolasa a pH 5.
Las enfermedades lisosomales de almacenamiento son un grupo de más de 50 enfermedades genéticas recesivas causadas por déficit de una o más hidrolasas lisosomales. Sin la enzima funcional, el sustrato no digerido se acumula en el lisosoma, formando cuerpos residuales que crecen progresivamente y alteran la función celular.
- Enfermedad de Pompe: déficit de α-1,4-glucosidasa lisosomal. El glucógeno no puede ser degradado y se acumula en músculo e hígado. Produce miopatía y cardiomiopatía.
- Enfermedad de Tay-Sachs: déficit de hexosaminidasa A. Se acumulan gangliósidos GM2 en neuronas. Los niños afectados presentan deterioro neurológico progresivo y fallecen antes de los 5 años.
- Enfermedad celular-I (I-cell disease): déficit de N-acetilglucosamina fosfotransferasa. Sin esta enzima, las hidrolasas lisosomales no son marcadas con M6P y se secretan al exterior en lugar de dirigirse al lisosoma. Los lisosomas quedan vacíos de enzimas y acumulan material no digerido. Las enzimas lisosomales aparecen en sangre pero no actúan por el pH neutro.
| Enfermedad | Enzima deficitaria | Sustrato acumulado | Manifestación principal |
|---|---|---|---|
| Enfermedad de Pompe (glucogenosis tipo II) | α-1,4-glucosidasa lisosomal (maltasa ácida) | Glucógeno | Miopatía · cardiomiopatía · insuficiencia respiratoria |
| Enfermedad de Tay-Sachs | Hexosaminidasa A | Gangliósido GM2 | Deterioro neurológico progresivo · ceguera · muerte antes de los 5 años |
| Enfermedad de Gaucher | Glucocerebrosidasa | Glucocerebrósido | Hepatoesplenomegalia · afectación ósea · anemia (tipo 1: sin afectación neurológica) |
| Enfermedad de Niemann-Pick | Esfingomielinasa (tipo A/B) | Esfingomielina | Hepatoesplenomegalia · afectación neurológica (tipo A) |
| Mucopolisacaridosis (MPS) | Varias enzimas según el tipo | Glucosaminoglicanos | Dismorfia facial · hepatoesplenomegalia · afectación ósea y neurológica |
| Enfermedad celular-I (I-cell disease) | N-acetilglucosaminil fosfotransferasa | Múltiples sustratos (todas las hidrolasas se secretan) | Dismorfias graves · retraso psicomotor · muerte en infancia |
8.4. El peroxisoma: características generales
El peroxisoma fue descubierto en 1956 por Rhodin, que los denominó inicialmente «microcuerpos». Están presentes en todas las células eucariotas sin excepción.
Morfológicamente son vesículas esféricas de 0,15 a 0,5 μm de diámetro, rodeadas de una membrana simple. A diferencia de las mitocondrias y los cloroplastos, los peroxisomas no tienen ADN ni ribosomas: todas sus proteínas se importan del citosol. Contienen gran cantidad de enzimas oxidativos; los más abundantes son la catalasa y la urato oxidasa. La urato oxidasa está en tan alta concentración que puede precipitar formando una estructura cristalina denominada nucleoide, visible al microscopio electrónico.
8.5. Funciones del peroxisoma
8.5.1. Oxidación de sustratos y neutralización del H₂O₂
La función central del peroxisoma es la oxidación de sustratos utilizando O₂. Las reacciones producen peróxido de hidrógeno (H₂O₂) como subproducto:
RH₂ + O₂ → R + H₂O₂
El H₂O₂ es un oxidante potente y tóxico. La catalasa lo neutraliza de dos formas: usándolo para oxidar otros sustratos tóxicos (fenoles, formaldehído, alcohol etílico, ácido fórmico) o descomponiéndolo directamente en agua y oxígeno cuando se acumula en exceso:
2 H₂O₂ → 2 H₂O + O₂
De este modo el peroxisoma actúa como «compartimento de detoxificación»: neutraliza el H₂O₂ que genera y aprovecha su poder oxidante para eliminar otras sustancias tóxicas. El etanol, por ejemplo, se oxida a acetaldehído en el peroxisoma hepático.
8.5.2. β-oxidación de ácidos grasos
El peroxisoma realiza la β-oxidación de los ácidos grasos de cadena muy larga (más de 22 carbonos), que no pueden entrar directamente en la mitocondria. El peroxisoma los acorta hasta cadenas que pueden ser manejadas por la mitocondria, donde se completa la β-oxidación. El producto de la β-oxidación peroxisomal es acetil-CoA, que sale al citosol.
8.5.3. Síntesis de plasmalógenos
En células animales, el peroxisoma participa en la síntesis de plasmalógenos, un tipo de fosfolípidos especiales muy abundantes en la mielina del sistema nervioso y en las membranas de las células musculares cardíacas. Las alteraciones en la síntesis de plasmalógenos explican la afectación neurológica característica de muchas enfermedades peroxisomales.
8.6. Biogénesis del peroxisoma
La biogénesis del peroxisoma difiere fundamentalmente de la del lisosoma: los peroxisomas no reciben proteínas del RE ni del Golgi. Todas sus proteínas, tanto enzimas como proteínas de membrana, se sintetizan en ribosomas libres del citosol y son importadas al peroxisoma mediante señales específicas (PTS1 y PTS2, peroxisomal targeting signals) reconocidas por receptores de la membrana peroxisomal (peroxinas).
Los peroxisomas crecen incorporando proteínas y lípidos y se dividen por fisión cuando alcanzan un tamaño suficiente, generando dos peroxisomas hijos. Este mecanismo es análogo al de la división de las mitocondrias.
Las enfermedades peroxisomales son poco frecuentes pero graves, con afectación predominante del sistema nervioso por el déficit de plasmalógenos en la mielina.
- La enfermedad de Zellweger es la más grave: una mutación en una proteína integral de la membrana del peroxisoma impide la importación de proteínas peroxisomales. Los peroxisomas están presentes pero vacíos de enzimas. Los niños afectados presentan malformaciones cerebrales graves y fallecen en los primeros meses de vida.
- La adrenoleucodistrofia (ALD) es causada por el déficit del transportador peroxisomal de ácidos grasos de cadena muy larga. Su acumulación destruye la mielina del sistema nervioso central y la corteza suprarrenal. Es la enfermedad popularizada por la película "El aceite de Lorenzo".
| Característica | Lisosoma | Peroxisoma |
|---|---|---|
| Tamaño | 0,2–0,5 μm | 0,15–0,5 μm |
| pH interno | ~5 (ácido) | ~7 (neutro) |
| Enzimas principales | >50 hidrolasas ácidas | Oxidasas · catalasa · enzimas de β-oxidación |
| Función principal | Digestión intracelular de macromoléculas | Oxidación de sustratos tóxicos · β-oxidación de AGCML · síntesis de plasmalógenos |
| Origen de proteínas | RE → Golgi → endosoma tardío (vía M6P) | Ribosomas libres del citosol (importación directa) |
| Biogénesis | Maduración del endosoma tardío | Fisión de peroxisomas preexistentes |
| Subproducto | Productos de digestión (aminoácidos, monosacáridos) | H₂O₂ (neutralizado por catalasa) |
| Patología | Enfermedades de almacenamiento lisosomal | Síndrome de Zellweger · adrenoleucodistrofia |
