9.1. Características generales
La mitocondria es el único orgánulo visible al microscopio óptico además del núcleo. Tiene un diámetro de 0,5 a 1 μm y una longitud muy variable. Puede visualizarse en células vivas con colorantes específicos como el verde Jano (que reacciona con la citocromo oxidasa) o fluorocromos como la rodamina 123, que se acumula en la mitocondria por su potencial de membrana negativo.
9.1.1. Localización y número
Las mitocondrias se distribuyen por todo el citoplasma, pero su localización no es aleatoria: se concentran donde más se necesita ATP. En las células del túbulo contorneado renal se acumulan en las invaginaciones basales, junto a las bombas de Na⁺/K⁺ que consumen gran cantidad de ATP. En el espermatozoide se localizan en la pieza intermedia, envueltas en espiral alrededor del axonema del flagelo para proporcionar la energía necesaria para la natación.
El número varía enormemente según el tipo celular y su actividad metabólica: desde unas pocas decenas en células poco activas hasta más de 100.000 en el ovocito. En el organismo humano, la mayoría de las células tienen cientos o miles de mitocondrias. Lo que importa fisiológicamente no es el número absoluto sino el volumen mitocondrial en relación al volumen celular.
Las mitocondrias son orgánulos dinámicos: se mueven por el citoplasma guiadas por los microtúbulos, cambian de forma y pueden fusionarse entre sí para formar redes mitocondriales más grandes o fisionarse para generar mitocondrias individuales.
9.2. Ultraestructura
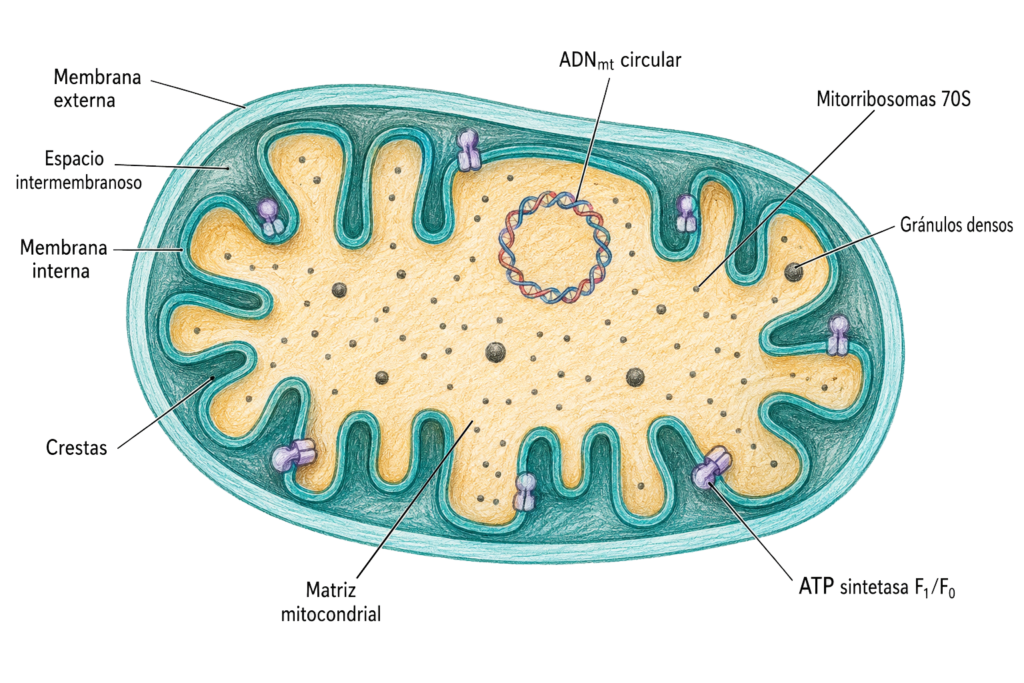
La mitocondria tiene doble membrana, lo que genera cuatro compartimentos distintos con funciones específicas:
9.2.1. Membrana externa
La membrana externa rodea completamente la mitocondria. Es relativamente permeable gracias a la presencia de porinas (proteínas de canal denominadas VDAC, Voltage-Dependent Anion Channel) que forman poros acuosos que permiten el paso libre de moléculas de hasta 5.000 Da: iones, nucleótidos, metabolitos pequeños.
Su composición es: 40% lípidos (fosfatidilcolina, fosfatidiletanolamina, poco colesterol) y 60% proteínas. Además de las porinas, contiene enzimas del metabolismo lipídico.
9.2.2. Espacio intermembranoso
El espacio entre la membrana externa y la interna tiene una composición similar al citosol, ya que las porinas permiten el paso libre de moléculas pequeñas. Contiene enzimas específicas como la adenilquinasa y el citocromo c, una proteína soluble que participa en la cadena respiratoria y que, cuando se libera al citoplasma, activa la vía intrínseca de la apoptosis.
9.2.3. Membrana interna
La membrana interna es la más especializada de todas las membranas celulares. Su composición es: 80% proteínas y 20% lípidos, la relación más alta de cualquier membrana biológica. No contiene colesterol y es rica en cardiolipina, un fosfolípido exclusivo de las membranas bacterianas y mitocondriales que contribuye a la impermeabilidad de la membrana.
Esta impermeabilidad es esencial para la función bioenergética: la membrana interna es impermeable a prácticamente todos los iones y moléculas excepto a través de transportadores específicos. El gradiente de protones generado por la cadena respiratoria solo puede aprovecharse si la membrana es impermeable a los H⁺.
La membrana interna forma numerosas invaginaciones hacia la matriz denominadas crestas, que aumentan su superficie hasta 5 veces la de la membrana externa. La morfología de las crestas varía: en la mayoría de células son transversales (perpendiculares al eje mayor); en algunas son tubulares. En la membrana interna se localizan los cuatro complejos de la cadena respiratoria y la ATP sintetasa:
- Complejo I (NADH deshidrogenasa): más de 40 subunidades.
- Complejo III (citocromo b-c₁): dímero, cada monómero con 11 subunidades.
- Complejo IV (citocromo oxidasa): dímero, cada monómero con 13 subunidades.
- ATP sintetasa (complejo F₁/F₀): ~500 kDa, sintetiza ATP a partir del gradiente de protones.
Además, contiene numerosos transportadores específicos: ADP/ATP, fosfato, ácidos dicarboxílicos, ácidos tricarboxílicos, aminoácidos, ácidos grasos y calcio.
| Característica | Membrana externa | Membrana interna |
|---|---|---|
| Composición proteína/lípido | 60% proteínas · 40% lípidos | 80% proteínas · 20% lípidos |
| Colesterol | Presente (pequeña cantidad) | Ausente |
| Lípido especial | Fosfatidilcolina · fosfatidiletanolamina | Cardiolipina (exclusiva de bacterias y mitocondrias) |
| Permeabilidad | Permeable a moléculas <5.000 Da (porinas/VDAC) | Impermeable a iones y la mayoría de moléculas |
| Proteínas características | Porinas (VDAC) · enzimas del metabolismo lipídico | Complejos I, III, IV de la cadena respiratoria · ATP sintetasa · transportadores específicos |
| Morfología | Lisa | Muy plegada en crestas (5× más superficie que la externa) |
| Función | Barrera permeable · metabolismo lipídico | Fosforilación oxidativa · gradiente de protones |
9.2.4. Matriz mitocondrial
La matriz es el compartimento más interno, rodeado por la membrana interna. Contiene:
- ADN mitocondrial (ADNmt): de doble cadena, circular y cerrado. Tiene 16.569 pares de bases y codifica 13 polipéptidos (todos subunidades de los complejos respiratorios y la ATP sintetasa), 22 ARNt y 2 ARNr (12S y 16S). El código genético mitocondrial difiere ligeramente del código universal.
- Ribosomas mitocondriales (mitoribosomas): de tipo 70S, similares a los ribosomas procariotas.
- ARNt mitocondriales: los 22 ARNt codificados por el ADNmt son suficientes para traducir todos los codones mitocondriales.
- Enzimas del ciclo de Krebs y de otras rutas metabólicas.
- Gránulos densos: depósitos de proteínas y calcio visibles al microscopio electrónico.
| Característica | ADN mitocondrial | ADN nuclear |
|---|---|---|
| Forma | Circular · doble cadena · cerrado | Lineal · doble cadena |
| Tamaño | 16.569 pb | ~3.200 Mb (haploide) |
| Número de copias por célula | Cientos a miles (múltiples por mitocondria) | 2 (diploide) · 1 (haploide en gametos) |
| Proteínas asociadas | Sin histonas | Con histonas (nucleosomas) |
| Lo que codifica | 13 polipéptidos · 22 ARNt · 2 ARNr (12S y 16S) | ~20.000 genes proteicos + ARN no codificante |
| Código genético | Ligeramente distinto al universal | Universal |
| Herencia | Exclusivamente materna | Biparental (autosómica) · materna (ligada al X en hembras) · paterna (ligada al Y) |
| Reparación del ADN | Mecanismos poco eficaces | Múltiples sistemas de reparación eficaces |
| Protección frente a radicales libres | Escasa (sin histonas · próximo a la cadena respiratoria) | Alta (histonas · alejado de la cadena respiratoria) |
9.3. Funciones de la mitocondria
9.3.1. Producción de ATP
La función principal de la mitocondria es la síntesis de ATP mediante la fosforilación oxidativa. El proceso ocurre en dos fases:
- En la matriz se producen la formación de acetil-CoA (a partir de piruvato, ácidos grasos y aminoácidos) y el ciclo de Krebs, que oxida el acetil-CoA generando NADH, FADH₂ y GTP.
- En la membrana interna, los electrones del NADH y FADH₂ recorren la cadena respiratoria (complejos I, III y IV), reduciendo finalmente el O₂ a H₂O. La energía liberada en este proceso se usa para bombear H⁺ al espacio intermembranoso. El gradiente de protones resultante impulsa la ATP sintetasa (complejo F₁/F₀), que sintetiza ATP a partir de ADP y Pi.
9.3.2. Otras funciones metabólicas
La mitocondria también participa en la neoglucogénesis, la síntesis de ácidos grasos, el metabolismo de aminoácidos y el ciclo de la urea (ureogénesis).
9.3.3. Participación en la apoptosis
La mitocondria es el centro de control de la vía intrínseca de la apoptosis. Ante señales de daño celular irreparable (daño en el ADN, estrés oxidativo, falta de factores de supervivencia), las proteínas proapoptóticas de la familia Bcl-2 permeabilizan la membrana externa mitocondrial y permiten la liberación del citocromo c al citoplasma. El citocromo c forma el apoptosoma con Apaf-1 y activa la caspasa-9, desencadenando la cascada de caspasas ejecutoras.
La apoptosis, sus vías intrínseca y extrínseca, y su comparación con la necrosis se desarrollan en T29 — Envejecimiento y muerte celular.
9.4. Origen endosimbiótico de la mitocondria
La teoría endosimbiótica (Lynn Margulis, 1967) propone que las mitocondrias derivan de bacterias aerobias que fueron fagocitadas por una célula eucariota ancestral hace aproximadamente 1.500 millones de años, estableciendo una relación simbiótica en lugar de ser digeridas.
Los argumentos que sustentan esta teoría son:
- Doble membrana: la membrana interna es equivalente a la membrana plasmática bacteriana; la externa deriva de la membrana de la vesícula de fagocitosis.
- ADN circular de doble cadena sin histonas, igual que el ADN bacteriano.
- Ribosomas 70S sensibles a los mismos antibióticos que inhiben la síntesis proteica bacteriana (cloranfenicol, estreptomicina).
- División por fisión binaria, igual que las bacterias.
9.5. Biogénesis de la mitocondria
Las mitocondrias se originan exclusivamente por fisión de mitocondrias preexistentes: nunca se forman de novo. Una mitocondria crece incorporando nuevas proteínas y lípidos y se divide por fisión generando dos mitocondrias hijas con el mismo ADNmt.
Dado que el ADNmt solo codifica 13 proteínas, las aproximadamente 1.500 proteínas mitocondriales restantes están codificadas en el genoma nuclear, se sintetizan en ribosomas libres del citoplasma y son importadas a la mitocondria mediante secuencias señal específicas reconocidas por los complejos de translocación TOM (membrana externa) y TIM (membrana interna).
El ADNmt se hereda exclusivamente por vía materna: todas las mitocondrias del cigoto proceden del óvulo, ya que las pocas mitocondrias del espermatozoide son ubiquitinadas y degradadas tras la fecundación.
Las enfermedades mitocondriales son causadas por mutaciones en el ADNmt o en genes nucleares que codifican proteínas mitocondriales. El ADNmt es especialmente vulnerable porque carece de histonas protectoras, sus mecanismos de reparación son poco eficaces y está expuesto a los radicales libres generados por la cadena respiratoria.
Las enfermedades mitocondriales afectan preferentemente a tejidos con alta demanda energética: músculo, cerebro, corazón y riñón. Producen cuadros de miopatía mitocondrial, encefalopatía, acidosis láctica y oftalmoplejía (síndrome MELAS, MERRF, síndrome de Kearns-Sayre).
Al heredarse por vía materna, una madre afectada transmite la enfermedad a todos sus hijos, pero un padre afectado no la transmite a ninguno.
