26.1. El envejecimiento celular
26.1.1. Concepto
La duración de la vida de una célula no es ilimitada. Las células somáticas tienen una capacidad de división finita. Tras un número determinado de ciclos proliferativos, pierden la capacidad de dividirse y entran en un estado de parada permanente denominado senescencia replicativa. La célula senescente permanece metabólicamente activa durante un tiempo en fase G0, pero ya no vuelve a dividirse y acaba muriendo.
Envejecimiento celular (senescencia replicativa): estado en el que una célula ha perdido irreversiblemente su capacidad de dividirse, permanece viable pero en parada permanente del ciclo celular, y acaba muriendo.
El envejecimiento celular no implica necesariamente muerte inmediata. Y a la inversa, la muerte celular no implica necesariamente envejecimiento previo. Por ejemplo, las neuronas que mueren durante el desarrollo embrionario son células jóvenes que han realizado pocas divisiones.
26.1.2. Manifestaciones morfológicas del envejecimiento celular
Las células senescentes presentan cambios morfológicos reconocibles:
- Aumentan de tamaño tanto el citoplasma como el núcleo.
- Se aplanan.
- Adoptan formas irregulares.
- Pierden contacto con las células vecinas.
Según el tipo celular, aparecen indicadores adicionales.
- En queratinocitos se acumulan citoqueratinas.
- En neuronas y miocitos cardíacos se acumula lipofuscina, un pigmento de desgaste autofluorescente.
- En hepatocitos aparece ceroide.
Estas sustancias se depositan en cuerpos residuales, vesículas que los lisosomas no consiguen degradar completamente y que quedan almacenadas en el citoplasma. La cantidad de cuerpos residuales en una célula es un indicador indirecto de su edad replicativa.
26.1.3. Las experiencias de Hayflick
A finales de los años 60, el microbiólogo Leonard Hayflick demostró experimentalmente que el envejecimiento celular es una propiedad intrínseca de la célula, no un artefacto de las condiciones de cultivo.
Trabajando con fibroblastos pulmonares humanos, Hayflick observó que los cultivos podían doblarse aproximadamente 50 veces antes de perder la capacidad proliferativa. Poco antes de alcanzar ese límite, las células mostraban los primeros signos de envejecimiento: tardaban más en confluir, crecían irregularmente. Este límite se conoce desde entonces como el límite de Hayflick.
Para demostrar que el límite residía en la célula y no en el medio de cultivo, diseñó una serie de experimentos adicionales:
- Mezcló fibroblastos masculinos con 40 doblajes realizados (con capacidad para 10 más) con fibroblastos femeninos con solo 10 doblajes realizados (capacidad para 40 más). Tras 20 doblajes adicionales, todas las células supervivientes eran femeninas: las masculinas habían agotado su límite exactamente cuando correspondía.
- Congeló células tras 20 y tras 40 doblajes. Al descongelarlas, pudieron realizar exactamente 30 y 10 doblajes más respectivamente, demostrando que el contador no se detiene con la congelación: la célula «recuerda» cuántas veces se ha dividido.
- Para localizar el factor regulador dentro de la célula, utilizó citochalasina B para expulsar núcleos celulares y obtener citoplastos (células sin núcleo). Al fusionar citoplastos jóvenes con células viejas, y citoplastos viejos con células jóvenes, el comportamiento de la célula fusionada seguía siempre al núcleo, no al citoplasma.
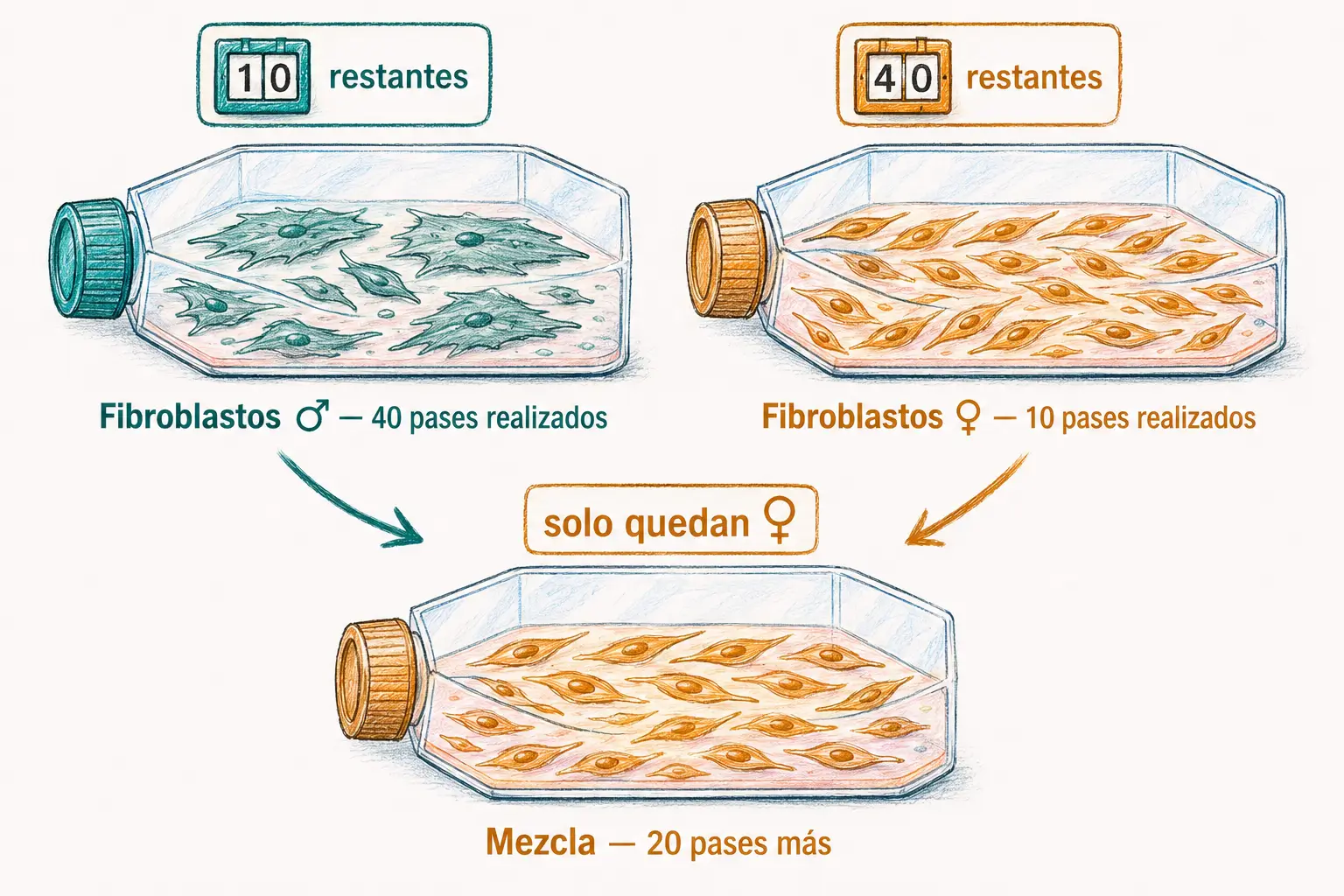
Las experiencias de Hayflick demuestran que el reloj del envejecimiento replicativo reside en el núcleo, no en el citoplasma. Debe existir, por tanto, un mecanismo genético que contabilice las divisiones.
26.1.4. Mecanismos del envejecimiento celular
El mecanismo más aceptado para explicar el límite de Hayflick es el acortamiento de los telómeros. En cada ronda de replicación del ADN, la síntesis de la hebra rezagada mediante fragmentos de Okazaki deja sin replicar el extremo 5′ del cromosoma, acortando ligeramente el telómero. Tras suficientes divisiones, los telómeros alcanzan una longitud mínima crítica que es detectada como daño en el ADN, lo que activa los mecanismos de control del ciclo celular y detiene permanentemente la proliferación.
La enzima telomerasa compensa este acortamiento sintetizando nuevas repeticiones teloméricas (TTAGGG en humanos). Sin embargo, su actividad está muy restringida. Está presente en células madre y con frecuencia en células tumorales, pero es prácticamente nula en células somáticas adultas diferenciadas. Esta ausencia de telomerasa en la mayoría de células del organismo es la base molecular del reloj replicativo de Hayflick.
Telomerasa y cáncer
Las células tumorales frecuentemente reactivan la telomerasa, lo que les permite superar el límite de Hayflick y proliferar de forma indefinida. La inhibición de la telomerasa es una de las dianas terapéuticas en investigación oncológica. El síndrome de Werner y la progeria (síndrome de Hutchinson-Gilford) son enfermedades genéticas que aceleran el envejecimiento celular, asociadas a disfunciones en la reparación del ADN y el mantenimiento telomérico.
Junto al acortamiento telomérico, contribuyen al envejecimiento celular:
- La acumulación de daño oxidativo por radicales libres.
- La sobreexpresión progresiva de inhibidores de las CDK, que frenan el ciclo celular.
Los mecanismos moleculares del acortamiento telomérico, la replicación del extremo de los cromosomas y el papel de la telomerasa se desarrollan en detalle en el tema niveles estructurales de los ácidos nucleicos de Bioquímica y Biología Molecular.
26.2. Muerte celular: necrosis y apoptosis
Las células pueden morir de dos formas radicalmente distintas en su mecanismo, su morfología y sus consecuencias para el tejido.
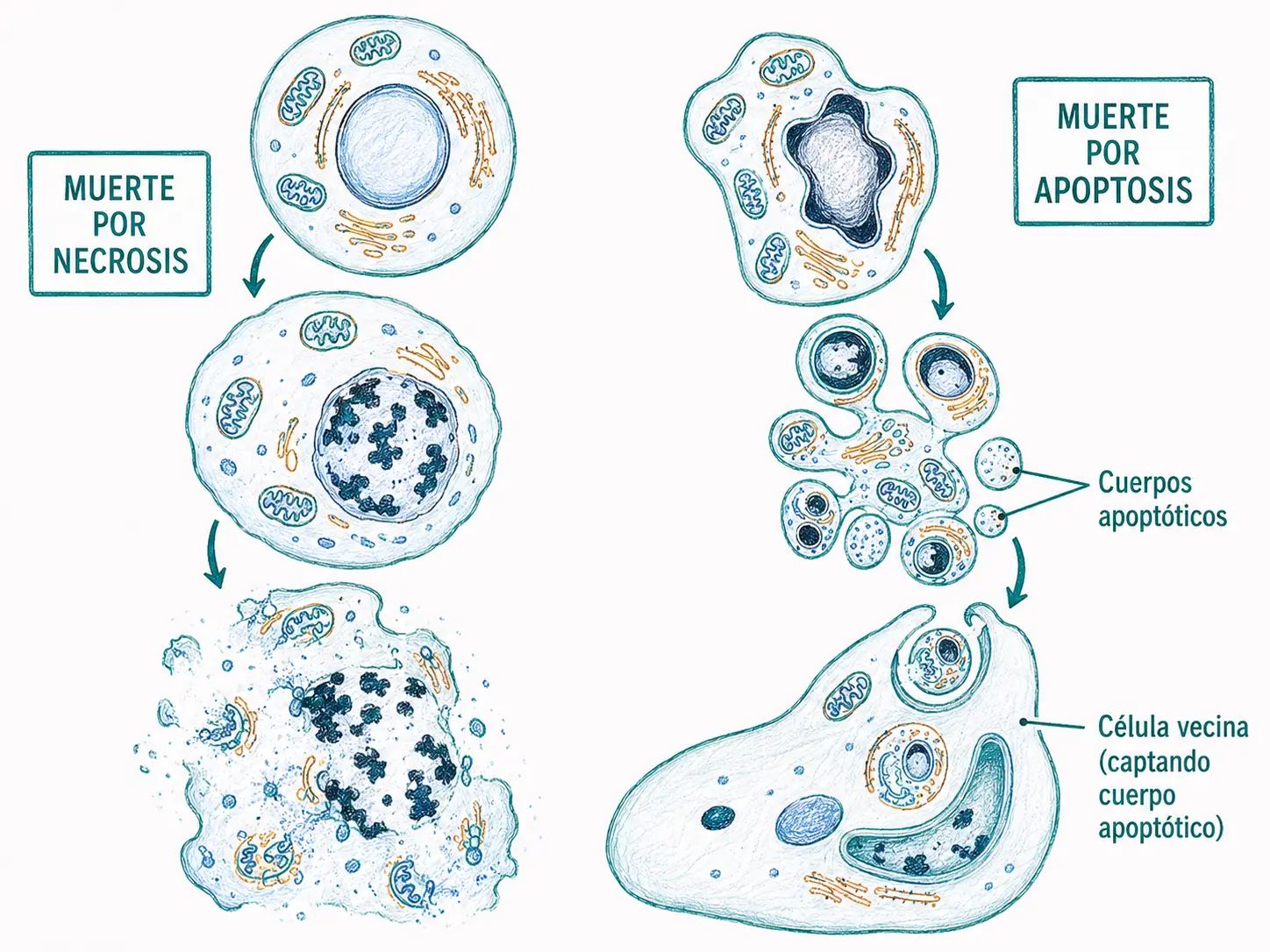
Necrosis: muerte celular accidental producida por agresiones externas (isquemia, tóxicos, traumatismos). Es pasiva, afecta a grupos de células y desencadena inflamación tisular.
Apoptosis: muerte celular programada, activa, regulada por un programa genético interno. Es fisiológica, afecta a células individuales y no produce inflamación.
26.2.1. Necrosis
La necrosis se produce cuando una agresión externa altera la función de la membrana plasmática, en particular la bomba Na⁺/K⁺-ATPasa. Al fallar el bombeo iónico, el gradiente osmótico arrastra agua hacia el interior celular:
- La célula se hincha, los orgánulos se alteran y finalmente la membrana se rompe, vertiendo el contenido intracelular al espacio extracelular.
- Este vertido activa la respuesta inflamatoria y requiere la acción de macrófagos y otras células especializadas para su eliminación.
Por estas razones, la necrosis siempre afecta simultáneamente a un grupo de células (todas las expuestas a la agresión), altera la estructura del tejido y puede comprometer la función del órgano.
26.2.2. Apoptosis
La apoptosis es la forma fisiológica de muerte celular. Es esencial durante el desarrollo embrionario (el 80% de las neuronas generadas en el embrión mueren por apoptosis para modelar el sistema nervioso), en el mantenimiento de la homeostasis tisular del adulto y en la eliminación de células dañadas o potencialmente tumorales.
A diferencia de la necrosis, la apoptosis se caracteriza por:
- Una contracción del citoplasma y del núcleo.
- Condensación y marginación de la cromatina hacia la periferia nuclear.
- Fragmentación de la célula en cuerpos apoptóticos: pequeñas vesículas rodeadas de membrana que encapsulan los restos celulares.
Los cuerpos apoptóticos son rápidamente fagocitados por células vecinas o por macrófagos sin que el contenido intracelular contacte con el espacio extracelular, evitando así la inflamación.
| Característica | Apoptosis | Necrosis |
|---|---|---|
| Origen | Programado, fisiológico | Accidental, patológico |
| Células afectadas | Individuales | Grupos de células |
| Volumen celular | Disminuye (contracción) | Aumenta (hinchazón) |
| Membrana plasmática | Intacta hasta el final | Se rompe |
| Orgánulos | Conservados | Alterados |
| Cromatina | Condensación regular, marginal | Condensación irregular |
| Resultado | Cuerpos apoptóticos fagocitados | Vertido al espacio extracelular |
| Inflamación | No | Sí |
| Alteración tisular | No | Sí |
26.3. Mecanismo de la apoptosis: las caspasas
El ejecutor molecular de la apoptosis es una familia de proteasas denominadas caspasas (cisteín-proteasas que escinden en residuos de ácido aspártico). Están presentes en todas las células animales, pero en forma de precursores inactivos denominados procaspasas.
La activación de las procaspasas ocurre por escisión proteolítica, generalmente llevada a cabo por otra caspasa ya activa. Una vez activas, las caspasas escinden y activan a su vez otras procaspasas, generando una cascada proteolítica amplificadora: unas pocas moléculas iniciadoras activan muchas caspasas efectoras, que desmantelan la célula de forma rápida e irreversible.
Las caspasas efectoras escinden sustratos que fragmentan las laminas nucleares (rotura de la lámina nuclear), liberan DNAsas que degradan el ADN en fragmentos de tamaño regular y desensamblan el citoesqueleto. El resultado es la destrucción ordenada y contenida de la célula.
La apoptosis es un mecanismo de todo o nada: una vez la célula supera el punto de activación de la cascada de caspasas, el proceso es irreversible. No hay vuelta atrás.
La apoptosis puede activarse por dos vías principales:
- Vía extrínseca: señales procedentes del exterior de la célula activan receptores de muerte en la membrana plasmática (como el receptor Fas). Esto recluta procaspasas iniciadoras (procaspasa-8) que se activan entre sí e inician la cascada. Los linfocitos T citotóxicos utilizan esta vía para eliminar células infectadas o tumorales.
- Vía intrínseca: señales de estrés o daño interno (daño en el ADN, hipoxia) inducen la liberación de citocromo c desde el espacio intermembrana de la mitocondria hacia el citosol. El citocromo c se une a la proteína adaptadora Apaf-1, que agrega y activa procaspasas iniciadoras (procaspasa-9), desencadenando la cascada.
Las bases moleculares de la cascada de caspasas, las proteínas de la familia Bcl-2 que regulan la permeabilidad mitocondrial y el papel de p53 en la activación de la apoptosis por daño en el ADN se estudian en profundidad en Bioquímica y Biología Molecular.
