27.1. Introducción: la célula enferma
La célula humana es un sistema altamente compartimentado. Cada orgánulo tiene una función definida, y cuando esa función falla, las consecuencias se propagan desde la escala molecular hasta la escala clínica. La patología celular estudia qué ocurre cuando un compartimento estructural se altera y cómo ese fallo se traduce en enfermedad.
Este tema es el cierre natural de la asignatura. A lo largo del curso has estudiado la membrana plasmática, los orgánulos, el citoesqueleto y el núcleo desde una perspectiva estructural y funcional normal. Ahora se aplica esa misma lógica en dirección contraria: si conoces la función, puedes deducir qué pasa cuando falla.
El patrón es siempre el mismo:
- El compartimento tiene una función.
- Una alteración genética, tóxica o metabólica perturba esa función.
- La célula intenta compensar.
- Si la compensación fracasa, aparece la enfermedad.
Este tema no repite los mecanismos de muerte celular (apoptosis, necrosis, envejecimiento celular), que ya se trataron en el Tema 26. Aquí se estudian las alteraciones estructurales de los compartimentos que preceden o acompañan a la enfermedad, no en el proceso final de la muerte.
27.2. Alteraciones de la membrana plasmática
27.2.1. Alteraciones de la bicapa lipídica
La composición lipídica de la membrana plasmática cambia con la temperatura, el estado metabólico y la edad celular. Cuando esa composición se altera de forma patológica, la fluidez de la membrana se compromete y con ella la función de las proteínas integrales que dependen de un entorno lipídico adecuado.
Un ejemplo clásico es la acumulación de colesterol en membranas de células envejecidas o dañadas, que reduce la fluidez y altera la señalización. En enfermedades como la aterosclerosis, la oxidación de lípidos de membrana en células endoteliales contribuye directamente al daño vascular.
La peroxidación lipídica es otro mecanismo importante. Los radicales libres de oxígeno atacan los ácidos grasos insaturados de la bicapa, generando productos tóxicos que alteran la permeabilidad y la integridad estructural de la membrana.
27.2.2. Alteraciones de las proteínas de membrana
Las proteínas de membrana pueden alterarse por:
- Mutación del gen que las codifica.
- Modificaciones post-traduccionales defectuosas.
- Degradación acelerada.
El resultado depende del tipo de proteína afectada. Si la proteína alterada es un canal iónico, el resultado es una canalopatía. El ejemplo más conocido es la fibrosis quística (mucoviscidosis), causada por mutaciones en el gen CFTR, que codifica un canal de cloro. La proteína mutada más frecuente (F508del) no se pliega correctamente y queda retenida en el RE en lugar de llegar a la membrana apical de las células epiteliales. El resultado es una secreción de moco espeso que obstruye los conductos pancreáticos, bronquiales e intestinales.
La fibrosis quística es la enfermedad genética grave más frecuente en población caucásica (1/2.500 nacidos vivos). La mutación F508del representa el 70% de los alelos patológicos. El diagnóstico precoz mediante cribado neonatal y los nuevos moduladores de CFTR (ivacaftor, lumacaftor) han transformado el pronóstico de esta enfermedad en las últimas dos décadas.
Si la proteína alterada es un receptor, el resultado puede ser resistencia a un ligando o señalización constitutiva. Las mutaciones con ganancia de función en receptores tirosina-quinasa son un mecanismo central en oncogénesis.
27.2.3. Alteraciones de las diferenciaciones de membrana
Las diferenciaciones de membrana, como microvellosidades, uniones estrechas o desmosomas, requieren una organización molecular precisa que involucra proteínas del citoesqueleto cortical. Su alteración tiene consecuencias directas en la función del epitelio.
La pérdida de uniones estrechas en el epitelio intestinal aumenta la permeabilidad paracelular, un fenómeno asociado a enfermedad inflamatoria intestinal y síndrome del intestino permeable. La alteración de los desmosomas, mediada por autoanticuerpos en el pénfigo vulgar, provoca la separación de los queratinocitos (acantólisis) y la formación de ampollas cutáneas graves.
La morfología microscópica de las diferenciaciones de membrana (microvellosidades, complejos de unión, hemidesmosomas) se estudia en detalle en Histología.
Este tema aborda únicamente las consecuencias patológicas de su alteración desde una perspectiva de Biología Celular. Puedes repasar el Tema 3 · Membrana celular II. Diferenciaciones y complejos de unión.
27.3. Alteraciones del retículo endoplasmático y el aparato de Golgi
27.3.1. Estrés del retículo endoplasmático y la respuesta UPR
El retículo endoplasmático rugoso (RER) es el lugar donde se pliegan las proteínas secretadas y de membrana. Para que el plegamiento sea correcto, el RE necesita un entorno específico: calcio, oxidación controlada y chaperonas moleculares como la BiP/GRP78.
Cuando se acumulan proteínas mal plegadas en el lumen del RE, se activa la respuesta a proteínas desplegadas o UPR (Unfolded Protein Response). La UPR es una respuesta adaptativa que actúa a tres niveles:
- Reduce la traducción global de proteínas para aliviar la carga del RE.
- Aumenta la expresión de chaperonas para ayudar al plegamiento.
- Activa la degradación de proteínas mal plegadas por el sistema ubiquitina-proteasoma (ERAD).
La UPR es inicialmente una respuesta de supervivencia. Solo cuando el estrés del RE es prolongado e irresoluble, la UPR activa señales proapoptóticas. La distinción entre UPR adaptativa y UPR proapoptótica depende de la duración e intensidad del estrés.
Si el estrés persiste y la UPR no logra restablecer la homeostasis del RE, se activa la apoptosis a través del factor de transcripción CHOP y de la caspasa-12. Este mecanismo está implicado en enfermedades neurodegenerativas (Parkinson, Alzheimer, ELA), en la diabetes tipo 2 (donde las células beta pancreáticas sobrecargan su RE con proinsulina) y en enfermedades hepáticas por acúmulo de proteínas mutadas como en el déficit de alfa-1 antitripsina.
27.3.2. Alteraciones del tráfico vesicular y el aparato de Golgi
El aparato de Golgi procesa y dirige las proteínas hacia su destino final, ya sea la membrana plasmática, los lisosomas o el exterior de la célula. Cuando el tráfico vesicular se altera, las proteínas no llegan a donde deben, con consecuencias diversas.
Un ejemplo ilustrativo es la enfermedad de inclusión de células I (I-cell disease o mucolipidosis tipo II). En este trastorno, una enzima del aparato de Golgi no añade el marcador manosa-6-fosfato a las hidrolasas lisosomales. Sin ese marcador, las enzimas no son dirigidas al lisosoma y se secretan al exterior de la célula en lugar de acumularse en el orgánulo. El resultado funcional es equivalente al de una enfermedad de depósito lisosomal: los lisosomas están vacíos de enzimas pero llenos de sustrato no digerido.
27.4. Alteraciones lisosomales y peroxisomales
27.4.1. Enfermedades por depósito lisosomal
Los lisosomas contienen más de 50 hidrolasas ácidas capaces de degradar prácticamente cualquier macromolécula biológica. Cuando una de esas enzimas falta o es defectuosa, su sustrato se acumula progresivamente en el interior del lisosoma. Estas son las enfermedades por depósito lisosomal (EDL), un grupo de más de 50 enfermedades genéticas raras.
En las enfermedades por depósito lisosomal, el fallo es enzimático pero la consecuencia es estructural: los lisosomas se llenan de material no digerido, se agrandan, y la función celular global se deteriora. La célula no puede "vaciar la papelera".
El patrón de herencia es autosómico recesivo en la mayoría de los casos. La clínica depende del tipo de sustrato acumulado y del tejido donde más se degrada. Las tres enfermedades más relevantes para el examen son:
| Enfermedad | Enzima deficiente | Sustrato acumulado | Tejido principalmente afectado |
|---|---|---|---|
| Gaucher | Glucocerebrosidasa | Glucocerebróxido | Macrófagos (hígado, bazo, médula ósea) |
| Niemann-Pick | Esfingomielinasa (tipo A/B) | Esfingomielina | Sistema nervioso, hígado, pulmón |
| Tay-Sachs | Hexosaminidasa A | Gangliósido GM2 | Neuronas (SNC) |
La enfermedad de Gaucher es la EDL más frecuente y también la que tiene tratamiento más establecido: la terapia de sustitución enzimática con glucocerebrosidasa recombinante (imiglucerasa) ha transformado su pronóstico.
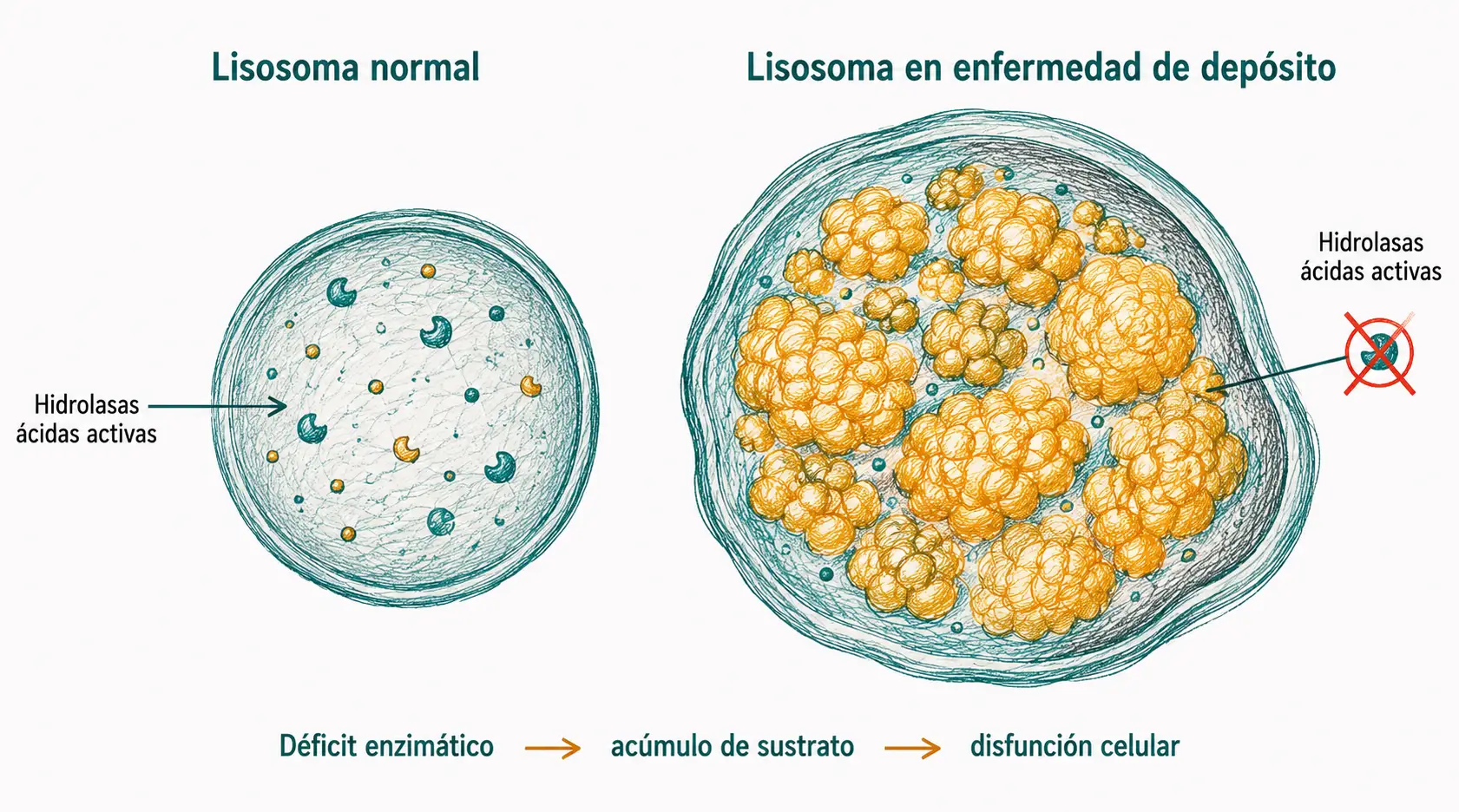
Las enfermedades de depósito lisosomal son enfermedades raras individualmente, pero colectivamente afectan a 1 de cada 5.000–7.500 nacidos vivos. Muchas son ahora tratables mediante terapia enzimática sustitutiva o chaperonas farmacológicas. El diagnóstico precoz es crítico porque el daño acumulado antes del diagnóstico puede ser irreversible, especialmente en el sistema nervioso.
27.4.2. Alteraciones de los peroxisomas
Los peroxisomas se encargan principalmente de la beta-oxidación de ácidos grasos de cadena muy larga y de la síntesis de plasmalógenos, lípidos esenciales para las vainas de mielina. Su alteración produce las enfermedades peroxisomales.
El síndrome de Zellweger (espectro Zellweger) es el más grave: se deben a defectos en la biogénesis del peroxisoma, de modo que el orgánulo no se forma correctamente. Los ácidos grasos de cadena muy larga se acumulan en plasma y tejidos, produciendo una disfunción neurológica grave, hipotonicidad y muerte en la primera infancia.
La adrenoleucodistrofia ligada al X (ALD) es otra enfermedad peroxisomal importante, causada por mutaciones en el transportador ABCD1, que introduce los ácidos grasos de cadena muy larga en el peroxisoma para su oxidación. Su acumulación destruye la mielina del SNC y la corteza suprarrenal.
27.5. Alteraciones mitocondriales
27.5.1. Disfunción mitocondrial y enfermedad
La mitocondria tiene dos roles críticos en la patología celular:
- Es la principal fuente de ATP.
- Es el centro de control de la apoptosis intrínseca
Cuando la mitocondria falla, la célula pierde energía y/o pierde el control sobre su propia supervivencia.
Las enfermedades mitocondriales en sentido estricto son las causadas por mutaciones que afectan a la cadena de transporte de electrones o a la fosforilación oxidativa. Los tejidos más afectados son siempre los que tienen mayor demanda energética: cerebro, músculo esquelético, corazón, retina y riñón.
La presentación clínica es variable y con frecuencia multisistémica, lo que dificulta el diagnóstico. Algunas formas tienen nombres propios reconocibles en el examen:
| Síndrome | Manifestaciones principales | Mutación típica |
|---|---|---|
| MELAS | Encefalomiopatía mitocondrial, acidosis láctica, episodios tipo ictus | ADNmt (m.3243A>G) |
| MERRF | Epilepsia mioclónica, fibras rojas rasgadas en biopsia muscular | ADNmt (m.8344A>G) |
| Leigh | Encefalopatía necrotizante subaguda, inicio en la infancia | ADNmt o ADN nuclear |
| Kearns-Sayre | Oftalmoplejia progresiva, bloqueo cardíaco, retinitis pigmentosa | Deleciones ADNmt |
27.5.2. ADN mitocondrial y herencia materna
El ADN mitocondrial (ADNmt) es circular, de doble cadena, y codifica 13 proteínas de la cadena respiratoria, 22 ARNt y 2 ARNr. Durante la fecundación, las mitocondrias del espermatozoide son eliminadas activamente en el zigoto; por tanto, todas las mitocondrias de un individuo provienen del óvulo materno.
Las enfermedades causadas por mutaciones en el ADNmt se heredan exclusivamente por vía materna. Una madre afecta transmite la enfermedad a todos sus hijos, pero ningún hijo varón la transmite a su descendencia.
Un concepto adicional importante es la heteroplasmia: dentro de una misma célula pueden coexistir mitocondrias con ADNmt normal y mutado. La proporción de mitocondrias mutadas determina en parte la gravedad clínica. Si la proporción de ADNmt mutado supera un umbral crítico (efecto umbral), aparece el fenotipo patológico.
No todas las enfermedades mitocondriales tienen herencia materna. Muchas proteínas mitocondriales están codificadas en el ADN nuclear y se heredan de forma mendeliana (autosómica recesiva o ligada al X). Solo las causadas por mutaciones en el propio ADNmt siguen la herencia materna estricta.
27.6. Alteraciones del citoesqueleto
27.6.1. Alteraciones de microtúbulos: ciliopatías
Los microtúbulos forman el axonema de cilios y flagelos, cuya estructura 9+2 permite el movimiento ciliar coordinado. Cuando las proteínas del axonema son defectuosas, los cilios no se mueven correctamente. Esta alteración define el grupo de las ciliopatías primarias.
La discinesia ciliar primaria (DCP) es el prototipo. Los cilios del epitelio respiratorio no barren el moco de forma eficaz, lo que produce infecciones respiratorias de repetición y bronquiectasias. Los espermatozoides con flagelos defectuosos no se desplazan, produciendo infertilidad masculina. Los cilios nodales, necesarios para establecer la asimetría izquierda-derecha del embrión, no funcionan, lo que en el 50% de los pacientes produce situs inversus (órganos en espejo).
La tríada de bronquiectasias + situs inversus + infertilidad masculina es el síndrome de Kartagener, que es una forma de discinesia ciliar primaria. Su base celular es la disfunción del axonema ciliar por mutaciones en las dineínas axonemales (DNAI1, DNAH5 y otras). El diagnóstico se confirma por microscopía electrónica del axonema o por análisis genético.
Otro grupo de ciliopatías afecta a los cilios primarios, que son cilios inmóviles presentes en casi todas las células del organismo y funcionan como antenas de señalización. Su disfunción produce el síndrome de Bardet-Biedl (obesidad, polidactilia, retinosis, insuficiencia renal) y la poliquistosis renal autosómica dominante, una de las enfermedades genéticas más frecuentes.
27.6.2. Alteraciones de microfilamentos de actina
La red de actina cortical mantiene la forma celular y es esencial para la migración, la citocinesis y la adhesión. Su desorganización patológica facilita la invasión tumoral. Las células cancerosas reorganizan su citoesqueleto de actina para formar podosomas e invadopodios, estructuras especializadas en la degradación de la matriz extracelular.
La esferocitosis hereditaria ilustra el papel de la actina en la integridad eritrocitaria. Mutaciones en proteínas del citoesqueleto cortical del eritrocito (espectrina, ankirina, banda 4.1) desestabilizan la red de actina submembranosa, provocando que los eritrocitos pierdan su forma bicóncava y adquieran forma esférica. Los esferocitos son menos deformables y son destruidos prematuramente en el bazo, produciendo anemia hemolítica.
27.6.3. Alteraciones de filamentos intermedios
Los filamentos intermedios confieren resistencia mecánica a la célula. Su composición varía según el tipo celular: queratinas en epitelios, vimentina en células mesenquimales, desmina en músculo, neurofilamentos en neuronas y laminas A/C en el núcleo (que se tratan en el apartado 28.7 de este tema).
Las mutaciones en queratinas 5 y 14, expresadas en el epitelio basal de la piel, producen epidermólisis bullosa simple (EBS). Ante un traumatismo mínimo, las células basales no soportan la tensión mecánica, se lisan y se forman ampollas intraepidérmicas. La gravedad clínica depende del tipo de mutación y de si afecta al dominio central helicoidal de la queratina.
La epidermólisis bullosa simple es la demostración más directa de que los filamentos de queratina son el sistema de resistencia mecánica del epitelio. Sin queratinas funcionales, la piel literalmente se desprende con el roce.
27.7. Alteraciones del núcleo
27.7.1. Alteraciones de la envoltura nuclear: laminopatías
La lámina nuclear es una malla de filamentos intermedios (laminas A, B y C) que tapiza la cara interna de la envoltura nuclear. Proporciona soporte mecánico al núcleo, ancla la cromatina periférica y regula la expresión génica al interactuar con regiones específicas del genoma (dominios asociados a lámina o LADs).
Las mutaciones en el gen LMNA, que codifica laminas A y C, producen un grupo heterogéneo de enfermedades llamadas laminopatías. Lo llamativo es que mutaciones en el mismo gen producen fenotipos clínicos completamente distintos:
| Laminopatía | Fenotipo principal | Mecanismo propuesto |
|---|---|---|
| Distrofia muscular de Emery-Dreifuss | Debilidad muscular, contracturas, bloqueo cardíaco | Fragilidad nuclear en células sometidas a estrés mecánico |
| Cardiomiopatía dilatada familiar | Insuficiencia cardíaca, arritmias | Fragilidad nuclear en cardiomiocitos |
| Progeria de Hutchinson-Gilford | Envejecimiento acelerado desde la infancia | Progerina (forma truncada de lamina A) desestabiliza la lámina |
| Lipodistrofia parcial familiar (Dunnigan) | Pérdida de grasa subcutánea, resistencia a insulina | Alteración de la regulación génica en adipocitos |
La progeria de Hutchinson-Gilford no se hereda: es casi siempre una mutación de novo en LMNA que produce una proteína truncada llamada progerina. Los niños afectados presentan envejecimiento acelerado extremo y fallecen en torno a los 13-14 años, principalmente por enfermedad cardiovascular. Es un modelo único para estudiar el envejecimiento celular normal.
27.7.2. Alteraciones de la cromatina y el ciclo celular en patología
Las alteraciones de la cromatina en el contexto de la enfermedad son fundamentalmente de dos tipos:
- Las que afectan a la estabilidad del genoma (inestabilidad genómica). La inestabilidad genómica es una característica central del cáncer. Incluye la aneuploidía (número anormal de cromosomas), las translocaciones cromosómicas, las amplificaciones génicas y las deleciones. Muchas de estas alteraciones son visibles en el núcleo en interfase como cambios en el tamaño y la forma nucleolar, o en metafase como aberraciones cromosómicas.
- Las que afectan a la regulación epigenética. Las alteraciones epigenéticas, como la hipermetilación de promotores de genes supresores de tumor o la modificación anormal de histonas, alteran el programa de expresión génica sin cambiar la secuencia del ADN. Son igual de relevantes para la patología que las mutaciones.
Los mecanismos moleculares de la regulación epigenética (metilación del ADN, modificaciones de histonas, ARN no codificantes) se estudian en Bioquímica y Biología Molecular. En este tema se menciona su relevancia patológica, pero el detalle mecanístico corresponde a esa asignatura.
27.8. Integración: del compartimento alterado a la enfermedad
Llegados a este punto, es útil ver el patrón de conjunto. Prácticamente cualquier enfermedad con base celular puede cartografiarse sobre un compartimento concreto:
- La membrana plasmática, cuando falla, produce canalopatías (CFTR, fibrosis quística) o enfermedades de adhesión (pénfigo, EBS).
- El retículo endoplasmático, cuando acumula proteínas mal plegadas, produce estrés crónico que puede desembocar en neurodegeneración o diabetes.
- Los lisosomas, cuando pierden una hidrolasa, acumulan sustrato y producen EDL de espectro clínico amplísimo.
- Las mitocondrias, cuando su cadena respiratoria falla, producen enfermedades multisistémicas con predilección por los tejidos de alta demanda energética.
- El citoesqueleto, cuando se altera, produce ciliopatías, fragilidad epitelial o facilita la invasión tumoral.
- El núcleo, cuando su lámina se desestructura, produce laminopatías de fenotipos sorprendentemente dispares.
Conocer la función normal de un compartimento celular permite predecir el tipo de enfermedad que producirá su alteración. La patología celular no es una lista de enfermedades que memorizar: es la lógica funcional de la célula aplicada al fallo.
Lo que une todos estos ejemplos es el mismo principio: la célula es un sistema integrado y la falla de una de sus partes no es un evento aislado. Las enfermedades lisosomales afectan la autofagia y la señalización de mTOR. La disfunción mitocondrial desregula la apoptosis. Las alteraciones del citoesqueleto afectan la migración y la señalización mecánica. Todo está conectado, y la frontera entre «patología de orgánulo X» y «enfermedad sistémica Y» es siempre porosa.
