17.1. Introducción: el sistema colinérgico
La acetilcolina (ACh) fue el primer neurotransmisor identificado. Otto Loewi demostró en 1921 que el nervio vago libera una sustancia química que frena el corazón, y Henry Dale identificó posteriormente esa sustancia como acetilcolina. El descubrimiento les valió el Nobel de Fisiología y Medicina en 1936.
Las neuronas que sintetizan y liberan acetilcolina se denominan neuronas colinérgicas. Se encuentran en tres territorios principales:
- Las motoneuronas α del asta anterior de la médula espinal, responsables de la contracción del músculo esquelético.
- Todas las neuronas preganglionares del sistema nervioso autónomo (simpático y parasimpático) y todas las neuronas posganglionares parasimpáticas.
- Numerosos núcleos del SNC: el núcleo basal de Meynert, el núcleo septal medial y los núcleos pedunculopontinos, entre otros.
La transmisión colinérgica tiene lugar en contextos muy distintos: la unión neuromuscular (el modelo clásico), los ganglios autónomos y múltiples circuitos del SNC. Aunque el neurotransmisor es siempre el mismo, el tipo de receptor y el efecto postsináptico varían radicalmente según la localización. Esta diversidad funcional hace del sistema colinérgico uno de los más relevantes farmacológicamente.
La organización del sistema nervioso autónomo y el papel de las neuronas colinérgicas posganglionares parasimpáticas se desarrollan en el Tema 14 — Sistema nervioso autónomo.
17.2. Síntesis, almacenamiento y degradación de la acetilcolina
17.2.1. Síntesis
La síntesis de acetilcolina ocurre en el citosol del terminal presináptico y es, con diferencia, la más sencilla entre los neurotransmisores de bajo peso molecular: se produce en un único paso enzimático.
Síntesis de acetilcolina Acetil-CoA + Colina → Acetilcolina (ACh) + CoA
Enzima: colina acetiltransferasa (ChAT)
Los dos sustratos proceden de fuentes distintas. La colina se obtiene principalmente de la dieta (fosfolípidos de membrana) y, en una proporción importante, de la recaptación de la propia colina liberada tras la degradación de la ACh. El acetil-CoA se genera en la mitocondria y es transportado al citosol para participar en la síntesis.
La presencia de ChAT en un terminal nervioso es el marcador bioquímico que identifica una neurona como colinérgica.
17.2.2. Almacenamiento vesicular
Una vez sintetizada en el citosol, la ACh no permanece libre: se introduce activamente en vesículas sinápticas mediante el transportador vesicular de acetilcolina (VAChT). Este transportador usa el gradiente de pH entre el citosol y el interior de la vesícula como fuerza motriz.
Las vesículas cargadas no forman una reserva uniforme. Se organizan en dos pools funcionales:
- El pool de liberación inmediata (o pool activo), formado por vesículas ancladas a la zona activa de la membrana presináptica y listas para fusionarse con el primer potencial de acción.
- El pool de reserva (o de reservorio), formado por vesículas asociadas al citoesqueleto que se movilizan cuando la demanda supera la capacidad del pool activo.
Tras la exocitosis, la membrana vesicular se recupera por endocitosis mediada por clatrina o por el mecanismo kiss-and-run, se recarga de ACh mediante VAChT y vuelve a integrarse en el pool activo. Este reciclaje de membrana vesicular es indispensable para mantener la capacidad de liberación durante actividad sináptica sostenida.
El mecanismo de reciclaje vesicular por endocitosis se introduce como principio general en el Tema 16. En la unión neuromuscular cobra especial importancia porque la frecuencia de estimulación puede ser muy alta y el pool activo se agotaría rápidamente sin reposición.
17.2.3. Liberación dependiente de calcio
La llegada de un potencial de acción al terminal presináptico desencadena una secuencia de eventos que culmina en la exocitosis de ACh. El paso crítico es la entrada de calcio.
La despolarización de la membrana presináptica abre canales de calcio voltaje-dependientes (Cav), concentrados en las zonas activas. La concentración de Ca²⁺ en el citosol aumenta localmente de forma muy rápida e intensa: pasa de ~100 nM en reposo a valores que pueden superar los 100 µM justo bajo la membrana presináptica.
Este aumento de Ca²⁺ activa las proteínas SNARE, que median la fusión de la vesícula con la membrana presináptica y la apertura del poro de fusión. El contenido vesicular se vierte en la hendidura sináptica.
La liberación de ACh es estrictamente dependiente de Ca²⁺. Sin entrada de calcio no hay exocitosis, aunque el potencial de acción llegue con normalidad al terminal. El Ca²⁺ es el acoplador entre la señal eléctrica y la señal química.
Cada vesícula contiene aproximadamente 10.000 moléculas de ACh. Ese contenido constituye un cuanto: la unidad mínima de liberación. El concepto de liberación cuántica, formulado por Katz y Del Castillo en los años 50, se aplica directamente al modelo de la unión neuromuscular y se desarrolla en el apartado 17.4.
Incluso en reposo, sin ningún potencial de acción, se producen fusiones espontáneas de vesículas individuales. Cada una genera en el músculo una pequeña despolarización postsináptica llamada potencial en miniatura de placa motora (mEPP). Los mEPP tienen amplitud constante (~0,5 mV) porque reflejan la liberación de un único cuanto. Fueron la evidencia experimental que permitió a Katz y Del Castillo demostrar que la transmisión sináptica es cuántica, no continua.
17.2.4. Degradación y recaptación de colina
La terminación de la señal colinérgica depende de la hidrólisis enzimática de la ACh en la hendidura sináptica. La enzima responsable es la acetilcolinesterasa (AChE), una de las enzimas más rápidas del organismo: puede hidrolizar hasta 25.000 moléculas de ACh por segundo.
La reacción produce colina y acetato. La colina es recaptada activamente por el terminal presináptico mediante el transportador de alta afinidad CHT1 y se reutiliza para sintetizar nueva ACh. El acetato difunde libremente.
El ciclo de la acetilcolina es un ciclo cerrado: la colina liberada por la hidrólisis vuelve al terminal y se reutiliza. Aproximadamente el 50% de la colina de cada vesícula nueva procede de este reciclaje. La disponibilidad de colina es, por tanto, un factor limitante de la síntesis de ACh.
Este mecanismo de degradación se desarrolla con más detalle en el contexto de la unión neuromuscular (17.4.4), donde la AChE tiene una localización anatómica específica y una relevancia clínica y toxicológica directa.
17.3. Receptores de acetilcolina
La acetilcolina actúa sobre dos familias de receptores con propiedades moleculares y funcionales muy distintas. Aunque comparten el mismo ligando, sus mecanismos de acción son opuestos en velocidad y naturaleza.
- Receptor nicotínico: receptor ionotrópico de la acetilcolina, activado también por la nicotina. Forma un canal iónico pentamérico que se abre directamente al unirse el ligando.
- Receptor muscarínico: receptor metabotrópico de la acetilcolina, activado también por la muscarina (extraída de Amanita muscaria). Actúa a través de proteínas G, con respuestas más lentas y duraderas.
La distinción entre ambos tipos tiene consecuencias directas sobre dónde y cómo actúa la acetilcolina en el organismo, y explica por qué un mismo neurotransmisor puede producir efectos tan distintos según el tejido.
17.4.1. Receptores nicotínicos
Los receptores nicotínicos son canales iónicos regulados por ligando. Están formados por cinco subunidades proteicas transmembrana dispuestas simétricamente alrededor de un poro central: dos subunidades α, una β, una δ y una ε (en el músculo adulto; en el feto y en las neuronas la composición varía).
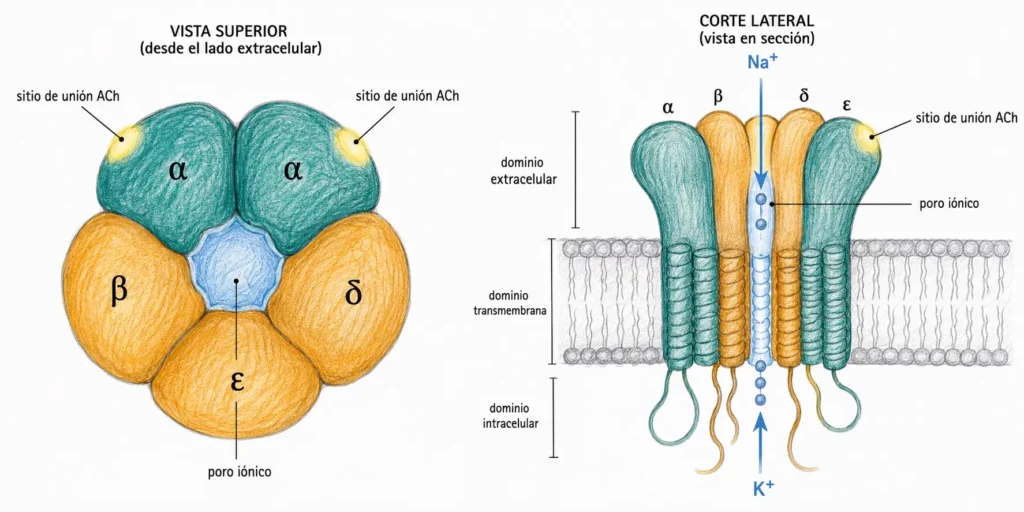
El canal es permeable a Na⁺, K⁺ y Ca²⁺. Cuando se abre, entran Na⁺ y Ca²⁺ por gradiente electroquímico y sale K⁺. El flujo neto de cargas positivas hacia el interior produce una despolarización de la membrana postsináptica.
Para que el canal nicotínico se abra es necesario que dos moléculas de ACh se unan simultáneamente, una a cada subunidad α. Sin las dos uniones el canal permanece cerrado. Esta cooperatividad de doble unión explica la sensibilidad del receptor a la concentración local de ACh.
Los receptores nicotínicos se encuentran en tres localizaciones principales: la placa motora terminal del músculo esquelético, los ganglios del sistema nervioso autónomo (simpático y parasimpático) y algunas neuronas del SNC. En los tres contextos la respuesta es una despolarización rápida, aunque la composición de subunidades varía.
Los receptores nicotínicos no son exclusivos de la unión neuromuscular. También están en los ganglios autónomos, donde median la transmisión entre la neurona preganglionar y la posganglionar, independientemente de si el ganglio es simpático o parasimpático.
Farmacología de los receptores nicotínicos
El agonista prototípico es la nicotina. El antagonista clásico en la placa motora es el curare y sus derivados sintéticos (vecuronio, rocuronio): compiten con la ACh por las subunidades α sin activar el canal, bloqueando la transmisión neuromuscular.
Los bloqueantes neuromusculares no despolarizantes (vecuronio, rocuronio, cisatracurio) son antagonistas competitivos de los receptores nicotínicos de la placa motora. Se usan en anestesia para producir relajación muscular controlada. Su efecto se revierte con inhibidores de la AChE (neostigmina) o con agentes quelantes específicos (sugammadex para el rocuronio).
17.4.2. Receptores muscarínicos
Los receptores muscarínicos son receptores acoplados a proteínas G (GPCRs). No forman canales por sí mismos: su activación desencadena cascadas de señalización intracelular que modulan la actividad celular de forma más lenta y sostenida.
Se dividen en cinco subfamilias (M1-M5) con segundos mensajeros, localizaciones y relevancia clínica distintas:
| Receptor | Proteína G / 2.º mensajero | Localización principal | Relevancia clínica |
|---|---|---|---|
| M1 | Gq → IP3/DAG → ↑ Ca²⁺ intracelular | SNC, ganglios del SNV, glándulas gástricas | Diana en enfermedad de Alzheimer (déficit colinérgico central) |
| M2 | Gi → ↓ AMPc; apertura de canales de K⁺ (GIRK) | Corazón (nodo sinusal, AV), terminales presinápticos colinérgicos (autorreceptor) | Media la bradicardia parasimpática; bloqueado por atropina en urgencias |
| M3 | Gq → IP3/DAG → ↑ Ca²⁺ intracelular | Músculo liso (bronquial, gastrointestinal, vesical), glándulas exocrinas | Broncoconstricción; diana de ipratropio y tiotropio en EPOC/asma |
| M4 | Gi → ↓ AMPc; cierre de canales de Ca²⁺ | SNC (estriado, córtex), terminales presinápticos | Modulación de la vía dopaminérgica; investigación en esquizofrenia |
| M5 | Gq → IP3/DAG → ↑ Ca²⁺ intracelular | SNC (sustancia negra, área tegmental ventral); muy escasos | Sin aplicación clínica establecida; en investigación |
Los receptores M1, M3 y M5 comparten la misma vía de señalización (Gq → IP3/DAG) y producen respuestas excitadoras. Los receptores M2 y M4 actúan a través de Gi, reduciendo el AMPc y produciendo respuestas inhibidoras. Esta división par/impar es útil para recordar la farmacología.
Los receptores muscarínicos pueden localizarse tanto en la postsinapsis como en la presinapsis de la propia neurona colinérgica, donde actúan como autorreceptores. Al detectar la ACh liberada, los subtipos M2 y M4 modulan hacia abajo la síntesis y la exocitosis de nuevas vesículas. En la presinapsis también se han descrito receptores M1 con efecto facilitador: la dualidad inhibidora (M2/M4) y facilitadora (M1) permite una regulación presináptica fina de la liberación de ACh según la concentración local del neurotransmisor.
Atropina: antagonista muscarínico no selectivo (bloquea M1-M5). Usos clínicos principales: bradicardia sintomática (bloquea M2 cardíaco), premedicación anestésica (reduce secreciones, bloquea M3 glandular) e intoxicación por organofosforados (revierte el exceso colinérgico muscarínico). A dosis altas produce el síndrome anticolinérgico clásico: sequedad de mucosas, midriasis, taquicardia, retención urinaria, confusión.
El papel de los receptores muscarínicos en la regulación de los órganos efectores autonómicos (corazón, músculo liso visceral y glándulas) se desarrolla en el T14 — Sistema nervioso autónomo.
17.4. La unión neuromuscular como modelo de sinapsis colinérgica
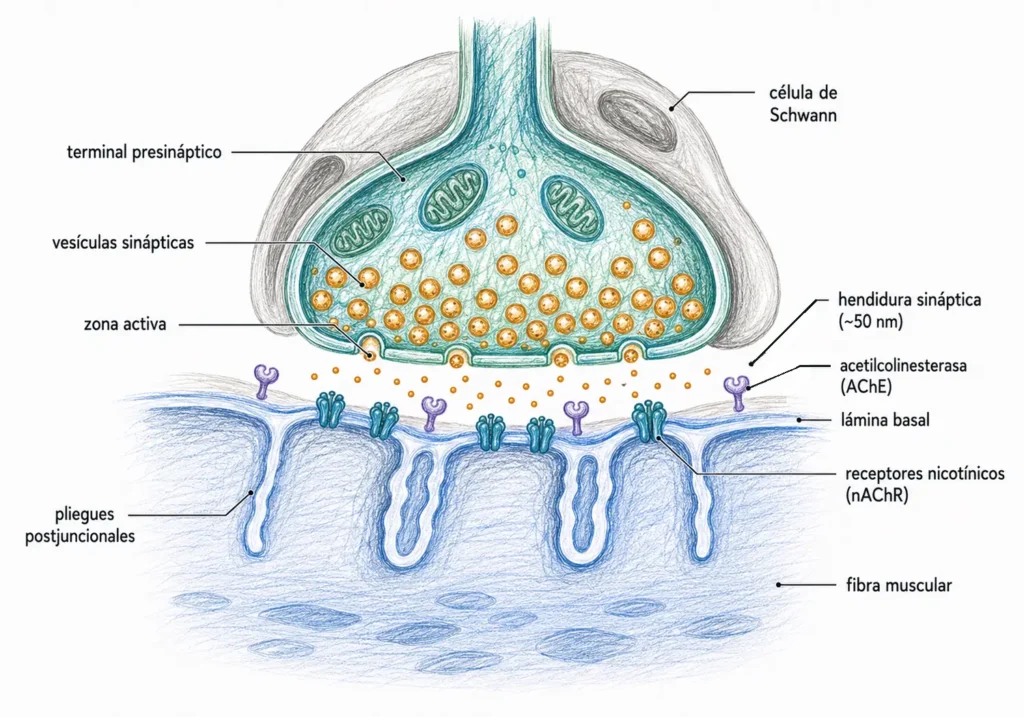
La unión neuromuscular es la sinapsis entre una motoneurona α y una fibra muscular esquelética. Es la sinapsis química mejor caracterizada y el modelo de referencia para entender la transmisión colinérgica: en este sistema se describieron por primera vez los cuantos de neurotransmisor, se identificó la AChE y se establecieron los principios del factor de seguridad sináptico.
17.4.1. Organización morfofuncional
Cada fibra muscular esquelética recibe inervación de una única motoneurona α. El terminal axónico se ramifica sobre la superficie muscular y cada rama forma una estructura especializada denominada placa motora terminal. El conjunto de todas las fibras inervadas por una misma motoneurona constituye una unidad motora.
La hendidura sináptica en la unión neuromuscular es más amplia que en las sinapsis del SNC (~50 nm frente a ~20 nm), lo que tiene consecuencias para la velocidad de difusión de la ACh y para la localización de la AChE.
La morfología detallada de la placa motora (invaginaciones postjuncionales, lámina basal especializada, células de Schwann pericapsulares) corresponde al alcance de Histología General. En Fisiología General interesa la función: qué ocurre desde que llega el potencial de acción hasta que se genera el potencial de placa motora.
La estructura microscópica de la unión neuromuscular se estudia en Histología General — Tejido muscular. La unidad motora y su papel en la graduación de la fuerza se retoman en el Tema 25 — Mecánica y electrofisiología muscular de esta asignatura.
17.4.2. Liberación por cuantos y factor de seguridad
El neurotransmisor de todas las uniones neuromusculares del músculo esquelético es la acetilcolina, sin excepción.
Cuando llega un potencial de acción al terminal presináptico, la entrada de Ca²⁺ a través de los canales Cav desencadena la fusión sincronizada de entre 100 y 200 vesículas por zona activa. Cada vesícula libera su cuanto de ~10.000 moléculas de ACh. En total, un único potencial de acción vierte en la hendidura del orden de un millón o más de moléculas de ACh.
La unión neuromuscular opera con un amplio factor de seguridad: se liberan aproximadamente tres veces más moléculas de ACh de las estrictamente necesarias para generar un potencial de acción en el sarcolema. Esto garantiza que cada impulso nervioso produzca invariablemente una contracción, incluso si parte de los receptores están bloqueados o reducidos.
Este margen tiene consecuencias clínicas directas: enfermedades como la miastenia gravis erosionan progresivamente el factor de seguridad antes de que la transmisión falle de forma clínicamente evidente.
Como se mencionó en 17.2.3, incluso en reposo se producen fusiones espontáneas de vesículas individuales que generan potenciales en miniatura de placa motora (mEPP). Su amplitud constante (~0,5 mV) refleja la liberación de un único cuanto y fue la evidencia experimental que permitió a Katz y Del Castillo establecer la naturaleza cuántica de la transmisión sináptica.
17.4.3. Activación del receptor y potencial de placa motora
La ACh liberada difunde a través de la hendidura sináptica y se une a los receptores nicotínicos concentrados en los pliegues de la placa motora. Como se describió en 17.3.1, la unión simultánea de dos moléculas de ACh a las subunidades α abre el canal. La entrada masiva de Na⁺ despolariza localmente el sarcolema, generando el potencial de placa motora (PPM).
El PPM es un potencial graduado, no un potencial de acción. Si su amplitud es suficiente para alcanzar el umbral en las zonas adyacentes del sarcolema, dispara un potencial de acción que se propaga por toda la fibra muscular e inicia la contracción.
La transmisión en la unión neuromuscular es de acción directa: la apertura del canal nicotínico genera la despolarización sin intermediarios de señalización intracelular. La secuencia completa, desde la llegada del potencial de acción a la motoneurona hasta el inicio de la contracción muscular, ocurre en apenas 5-6 ms.
El mecanismo por el que el potencial de acción en el sarcolema desencadena la contracción del sarcómero (acoplamiento excitación-contracción) se desarrolla en el Tema 24 — Contracción muscular.
17.4.4. Degradación de la acetilcolina: la acetilcolinesterasa
La terminación rápida de la señal en la placa motora depende de la hidrólisis de la ACh por la acetilcolinesterasa (AChE). En la unión neuromuscular, la AChE se presenta en su forma asimétrica: una molécula de gran tamaño anclada a la lámina basal de la hendidura sináptica mediante una subunidad de colágeno (ColQ). Esta localización estratégica garantiza que la ACh sea hidrolizada antes de poder difundir fuera de la sinapsis.
La velocidad de hidrólisis es muy elevada: la AChE puede degradar hasta 25.000 moléculas de ACh por segundo, lo que limita la duración de cada evento sináptico a pocos milisegundos. La colina liberada es recaptada por el terminal presináptico vía CHT1 y se reutiliza para sintetizar nueva ACh, cerrando el ciclo colinérgico descrito en 17.2.4.
Miastenia gravis: enfermedad autoinmune en la que se producen anticuerpos contra los receptores nicotínicos de la placa motora (principalmente contra la subunidad α). Los receptores funcionales disminuyen progresivamente, el factor de seguridad se erosiona y la transmisión neuromuscular se vuelve insuficiente. El resultado es fatiga muscular fluctuante que empeora con el ejercicio y mejora con el reposo.
El tratamiento de primera línea son los inhibidores de la AChE (piridostigmina): al impedir la degradación de la ACh, aumentan su concentración en la hendidura y compensan parcialmente la pérdida de receptores.
Los organofosforados (insecticidas como el paratión, agentes nerviosos como el sarín) inhiben la AChE de forma irreversible mediante fosforilación del sitio activo. La ACh se acumula en todas las sinapsis colinérgicas simultáneamente. El cuadro resultante combina efectos muscarínicos (broncoespasmo, hipersecreción, bradicardia, miosis) y nicotínicos (fasciculaciones, parálisis muscular).
El antídoto es atropina (bloquea los efectos muscarínicos) combinada con pralidoxima, cuya eficacia depende de la precocidad de la administración: la ventana varía entre pocas horas (agentes nerviosos como el sarín, con envejecimiento rápido de la fosforilación) y 24–48 horas (insecticidas organofosforados).
17.5. Otros contextos de la transmisión colinérgica
La unión neuromuscular es el modelo de referencia, pero la transmisión colinérgica opera en otros dos contextos igualmente importantes: los ganglios autónomos y el sistema nervioso central.
En los ganglios autónomos, la neurona preganglionar libera ACh que actúa sobre receptores nicotínicos de la neurona posganglionar, generando un potencial postsináptico excitador rápido (PPSE rápido). Este mecanismo es idéntico en el simpático y en el parasimpático. En algunos ganglios parasimpáticos existe además un PPSE lento mediado por receptores muscarínicos M1, que modula la excitabilidad posganglionar durante actividad sináptica sostenida. La diferencia entre ambos sistemas autónomos no está en la sinapsis ganglionar, sino en lo que ocurre después: el simpático usa noradrenalina sobre receptores adrenérgicos, y el parasimpático vuelve a usar ACh sobre receptores muscarínicos en el órgano efector.
En el SNC, la transmisión colinérgica participa en procesos de atención, memoria de trabajo y ciclo vigilia-sueño. Los núcleos colinérgicos más relevantes son el núcleo basal de Meynert, que proyecta al córtex y al hipocampo, y los núcleos colinérgicos del tronco del encéfalo. La degeneración de estas vías colinérgicas centrales es uno de los componentes del déficit cognitivo en la enfermedad de Alzheimer.
La organización del sistema nervioso autónomo, la transmisión ganglionar y los efectos posganglionares de la ACh muscarínica sobre los órganos efectores se desarrollan en el Tema 14 — Sistema nervioso autónomo.
El papel de las vías colinérgicas centrales en la cognición, la memoria y el ciclo vigilia-sueño se retoma en los temas de fisiología del sistema nervioso central.
