22.1. Generalidades de los neuropéptidos
Los neuropéptidos forman la familia más numerosa de neurotransmisores: en 1980 se conocían apenas 15-17; en 2000 se habían identificado más de 100. Su estudio fue inicialmente difícil porque actúan también como hormonas y neurohormonas, y porque nunca se liberan solos, sino en cotransmisión con otros neurotransmisores de molécula pequeña.
Todos tienen estructura peptídica, por lo que su actividad depende de su secuencia de aminoácidos. Se sintetizan como precursores inactivos de mayor tamaño que son procesados proteolíticamente para generar uno o varios péptidos activos. Un único gen precursor puede originar más de un neuropéptido activo, dependiendo del patrón de procesamiento en cada tejido.
Los neuropéptidos actúan predominantemente como neuromoduladores: no excitan ni inhiben directamente la neurona postsináptica, sino que modifican su sensibilidad al neurotransmisor principal que se libera de forma coexistente. Su efecto solo se manifiesta en condiciones biológicas específicas (dolor, saciedad, estrés), no en situaciones basales.
22.1.1. El principio de Dale y la cotransmisión
El principio de Dale en su formulación original planteaba que una neurona libera un solo neurotransmisor por todos sus terminales. La evidencia acumulada en las últimas décadas ha modificado esta idea: una neurona puede coexpresar y coLiberar simultáneamente uno o varios neurotransmisores de molécula pequeña junto con uno o varios neuropéptidos. Este fenómeno se denomina cotransmisión.
La cotransmisión es la regla en las sinapsis peptidérgicas. Los neuropéptidos actúan entonces como moduladores de la respuesta al neurotransmisor principal: amplifican, reducen o condicionan temporalmente su efecto sin sustituirlo.
La formulación correcta del principio de Dale revisado es: una neurona puede liberar múltiples neurotransmisores, pero siempre los mismos en todos sus terminales. No se viola que la neurona sea consistente en su identidad química; lo que se revisa es la restricción a un único mensajero.
22.2. Síntesis y mecanismo de liberación de los neuropéptidos
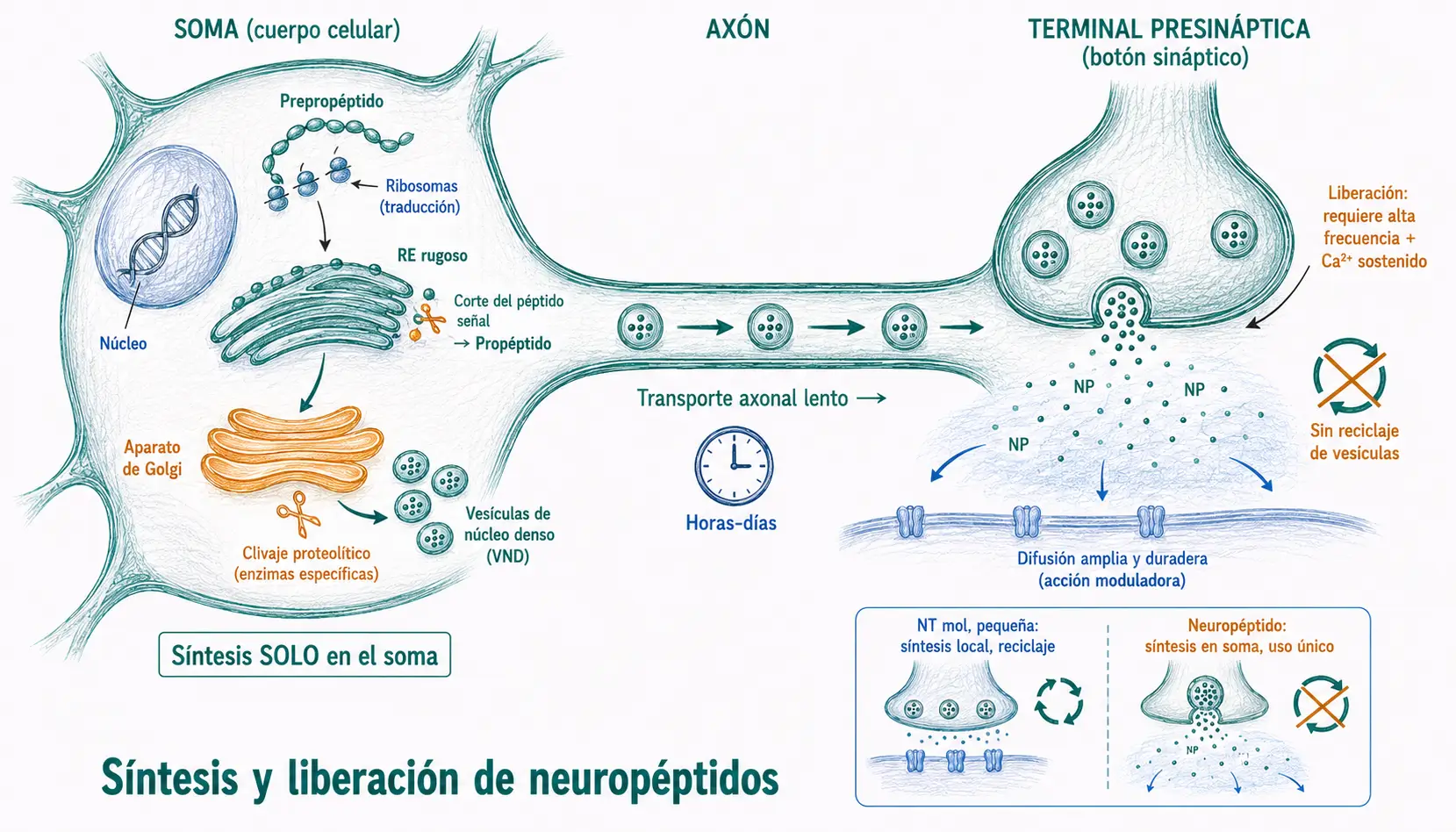
La síntesis de neuropéptidos sigue una ruta radicalmente distinta a la de los neurotransmisores de molécula pequeña, con consecuencias funcionales importantes:
Ruta de síntesis de los neuropéptidos:
- En el soma neuronal: los ribosomas sintetizan el prepropéptido (precursor inactivo con péptido señal).
- El prepropéptido entra en el retículo endoplasmático rugoso, donde se escinde el péptido señal y se obtiene el propéptido.
- El propéptido se empaqueta en vesículas de núcleo denso grande en el aparato de Golgi, donde continúa el procesamiento proteolítico hasta generar el péptido activo final.
- Las vesículas se transportan por transporte axonal lento desde el soma hasta la terminal presináptica.
- En la terminal, la liberación requiere estimulación de alta frecuencia y aumentos sostenidos de Ca²⁺ intracelular: condiciones más exigentes que las de los NT de molécula pequeña.
- Las vesículas de núcleo denso no se reciclan: se usan una sola vez y la reposición depende de nueva síntesis en el soma.
| Característica | Neuropéptidos | NT de molécula pequeña |
|---|---|---|
| Lugar de síntesis | Exclusivamente en el soma neuronal | En el terminal axónico |
| Vesícula | Vesícula de núcleo denso grande | Vesícula sináptica pequeña y clara |
| Transporte | Axonal lento desde soma hasta terminal | Síntesis local en el terminal |
| Liberación | Alta frecuencia, Ca²⁺ sostenido | Baja frecuencia, Ca²⁺ local |
| Reciclaje de vesículas | No: uso único | Sí: endocitosis y reutilización |
| Reposición | Lenta: depende de nueva síntesis en soma | Rápida: síntesis local inmediata |
La síntesis exclusivamente en el soma y el uso único de cada vesícula explican por qué los neuropéptidos se liberan solo ante estimulación intensa y sostenida. Una sinapsis peptidérgica activa en condiciones basales agotaría sus vesículas sin posibilidad de reponerlas localmente. Este diseño los convierte en señales de «alta demanda», no de transmisión basal.
22.3. Receptores de neuropéptidos
Todos los receptores de neuropéptidos son metabotrópicos acoplados a proteínas G, lo que implica respuestas lentas, duraderas y con capacidad de amplificación. No se ha identificado ningún receptor peptidérgico ionotrópico.
| Neuropéptido | Familia precursora | Receptores | Función principal |
|---|---|---|---|
| Endorfinas | POMC | μ (mu) | Analgesia, euforia, regulación hormonal |
| Encefalinas | Preproencefalina-A | μ (Met-Enk), δ (Leu-Enk) | Analgesia espinal, modulación del dolor |
| Dinorfinas | Preprodinorfina | κ (kappa) | Analgesia, disforia, efectos sedantes |
| Sustancia P | Preprosustancia P | NK1, NK3 | Transmisión nociceptiva, sensibilización central |
| CCK | Preprocolecistocinina | CCK-A, CCK-B | Saciedad, control de la ingesta |
| Vasopresina | Preproneurohipofisina II | V1, V2 | Vasoconstricción, reabsorción renal de agua |
| Oxitocina | Preproneurohipofisina I | OTR | Contracción uterina, eyección de leche, vinculación |
| VIP | Prepro-VIP | VPAC1, VPAC2 | Vasodilatación esplácnica, neuromodulación |
22.4. Principales neuropéptidos y sus funciones
22.4.1. Péptidos opiáceos
Se descubrieron investigando la existencia de ligandos endógenos de los receptores de morfina. Se han identificado más de 20, agrupados en tres familias según su gen precursor, con afinidades distintas por los tres tipos de receptores opioides (μ, δ, κ).
Los tres receptores opiáceos son GPCRs acoplados a proteína Gi, por lo que su activación inhibe la adenilato ciclasa (↓ AMPc), abre canales de K⁺ (hiperpolarización) y cierra canales de Ca²⁺ dependientes de voltaje en la presinapsis, reduciendo la liberación de neurotransmisor. El resultado neto es siempre inhibición neuronal y analgesia.
Las neuronas encefalinérgicas del asta posterior de la médula espinal controlan la entrada de información dolorosa al SNC: la liberación de encefalinas inhibe la transmisión del impulso nociceptivo antes de que ascienda por los haces espinotalámicos.
Morfina y opioides exógenos: actúan sobre los mismos receptores μ, δ y κ que los opioides endógenos. Su potente efecto analgésico reproduce y amplifica la acción de las encefalinas y las endorfinas. La tolerancia, la dependencia y el riesgo de sobredosis son consecuencia de la regulación a la baja de estos receptores (internalización y down-regulation) y de la supresión de la síntesis endógena ante la presencia crónica de opioides exógenos. Al retirar el opioide exógeno, el sistema carece transitoriamente tanto del fármaco como del opioide endógeno: esa es la base del síndrome de abstinencia.
22.4.2. Sustancia P
Fue el primer neuropéptido descubierto. Contiene 11 aminoácidos y se localiza en el hipocampo, la neocorteza y especialmente en el ganglio de la raíz dorsal, desde donde se libera en el asta posterior de la médula por estímulos dolorosos. Actúa sobre receptores NK1 (neurokinina 1) en la neurona de segundo orden y transmite información nociceptiva, térmica y autonómica periférica hacia el SNC.
Su liberación puede ser inhibida por opioides endógenos en la médula, suprimiendo la señal de dolor. Es también un hipotensor potente y participa en circuitos del control motor.
La sustancia P actúa en el asta posterior de la médula junto al glutamato para activar receptores NMDA y contribuir a la sensibilización central del dolor, un mecanismo compartido con el sistema glutamatérgico. Este proceso explica la alodinia y la hiperalgesia en el dolor crónico y se amplía en el contexto de los receptores NMDA espinales (Sinapsis glutamatérgicas).
22.4.3. Colecistocinina (CCK)
Se libera en los terminales del nervio vago en el núcleo del tracto solitario. Su función principal es el control de la ingesta: señaliza la saciedad al SNC, inhibiendo la conducta alimentaria. Es un ejemplo de neuropéptido cuya función principal es periférica (liberado por el intestino ante la presencia de grasas y proteínas en el duodeno) pero que actúa también centralmente como neuromodulador.
22.4.4. Vasopresina y oxitocina
Son neurohormonas sintetizadas en los núcleos supraóptico y paraventricular del hipotálamo a partir de precursores distintos pero estructuralmente relacionados. Actúan tanto como neurotransmisores/neuromoduladores en el SNC como como hormonas sistémicas liberadas desde la neurohipófisis.
La vasopresina (hormona antidiurética, ADH) regula la reabsorción de agua en el túbulo colector renal y produce vasoconstricción a dosis elevadas. La oxitocina induce las contracciones uterinas durante el parto, la eyección de leche en la lactancia y participa en el comportamiento de vinculación social y la respuesta al estrés.
La fisiología endocrina completa de la vasopresina y la oxitocina, incluyendo sus mecanismos de regulación hipotalámica, se estudia en el bloque de regulación sistémica de esta asignatura. En este tema se mencionan exclusivamente como ejemplo de neuropéptidos con función dual neuronal/endocrina.
22.4.5. Péptido intestinal vasoactivo (VIP)
Pertenece a la familia de las secretinas. Se localiza en el tubo digestivo, donde regula la vasodilatación de la región esplácnica. En el SNC actúa como neuromodulador potenciando los efectos de las catecolaminas. También está presente en neuronas de la corteza cerebral y en el núcleo supraquiasmático, donde participa en la regulación del ritmo circadiano.
22.5. Sinapsis purinérgicas: ATP y adenosina
Los sistemas purinérgicos no siguen el modelo clásico de sinapsis puntual. Sus componentes actúan en cascada: la degradación de unos genera otros, amplificando la señal con pocas moléculas. Por ello se habla de sistemas purinérgicos más que de sinapsis purinérgicas individuales.
Son de gran interés farmacológico, aunque su manipulación terapéutica es difícil precisamente por ese mecanismo en cadena.
22.5.1. Clasificación de los receptores purinérgicos
La nomenclatura actual (IUPHAR) organiza los receptores purinérgicos en dos grandes familias según el tipo de ligando:
| Familia | Ligando | Subtipos | Tipo de receptor | Efector principal |
|---|---|---|---|---|
| P1 | Adenosina | A1, A2A, A2B, A3 | Metabotrópico (GPCR) | Adenilato ciclasa (↑ o ↓ AMPc) |
| P2X | ATP | P2X1-7 | Ionotrópico (canal catiónico) | Na⁺/Ca²⁺ entra, K⁺ sale |
| P2Y | ATP, ADP, UTP, UDP | P2Y1, P2Y2, P2Y11-14 | Metabotrópico (GPCR) | PLC (↑ IP3/DAG) o adenilato ciclasa |
Los diadenosín polifosfatos (AP4A, AP5A, AP6A: dos adenosinas unidas por cadenas de fosfato) se liberan en cotransmisión con ATP y adenosina, y activan principalmente receptores P2. Existen en la literatura denominaciones antiguas como «receptores P4» para algunos de sus efectos, pero la nomenclatura IUPHAR actual los engloba dentro de la familia P2. No deben memorizarse como una familia independiente.
22.5.2. Colocalización y síntesis
Los sistemas purinérgicos se liberan casi siempre en cotransmisión. En las sinapsis noradrenérgicas, por ejemplo, se liberan aproximadamente 4 mol de noradrenalina por cada mol de ATP y adenosina. La pequeña cantidad de purinas liberada dificultó durante años el reconocimiento de su papel como neurotransmisores.
Al ser metabolitos de bajo peso molecular, el ATP y la adenosina se sintetizan en el terminal presináptico. Actúan como neurotransmisores solo si existe el transportador vesicular que los introduce en vesículas para su liberación cuántica.
En el SNC, los sistemas purinérgicos se concentran en zonas relacionadas con la actividad motora (cerebelo, ganglios basales). En el SNP existen nervios exclusivamente purinérgicos en vejiga, estómago y vasos deferentes.
22.5.3. Cascada de degradación purinérgica
La inactivación del ATP extracelular se produce por ecto-nucleotidasas ancladas en la cara externa de la membrana celular, que generan sucesivamente metabolitos con actividad propia sobre distintos receptores:
Cascada de degradación purinérgica:
ATP → ADP → AMP → Adenosina
ATP: activa receptores P2X (ionotrópicos) y P2Y (metabotrópicos)
ADP: activa receptores P2Y
AMP: sustrato de la 5'-nucleotidasa → genera adenosina
Adenosina: activa receptores P1 (A1, A2A, A2B, A3)
Enzimas implicadas:
ATP → ADP: ecto-ATPasa
ADP → AMP: ecto-ADPasa
AMP → adenosina: 5'-nucleotidasa
La adenosina resultante es recaptada por la presinapsis (vuelve a la ruta de síntesis de nucleótidos) o por la postsinapsis (sustrato metabólico).
La degradación en cadena del ATP no es solo inactivación: cada metabolito activa receptores distintos. Unas pocas moléculas de ATP liberadas pueden generar una respuesta amplificada y secuencial al activar sucesivamente P2X, P2Y y finalmente A1/A2A mediante la adenosina resultante. Este es el significado del «sistema en cadena» de la señalización purinérgica.
22.5.4. Receptores de adenosina (familia P1)
Los receptores de adenosina son los mejor caracterizados de los sistemas purinérgicos. Todos son GPCRs; se diferencian por su efector y su distribución:
| Receptor | Proteína G / efector | Efecto sobre AMPc | Mecanismo adicional | Localización principal |
|---|---|---|---|---|
| A1 | Gi → ↓ adenilato ciclasa | ↓ AMPc | Apertura de canales GIRK (K⁺) → hiperpolarización; cierre de canales Ca²⁺ presinápticos | SNC (hipocampo, corteza, cerebelo), corazón, tejido adiposo |
| A2A | Gs → ↑ adenilato ciclasa | ↑ AMPc | Vasodilatación, inhibición plaquetaria | SNC (estriado), vasos sanguíneos |
| A2B | Gs/Gq | ↑ AMPc / ↑ IP3 | Baja afinidad por adenosina; activo solo a concentraciones altas | Tejidos periféricos, intestino, pulmón |
| A3 | Gi → ↓ adenilato ciclasa | ↓ AMPc | Modulación de la respuesta inflamatoria | Tejidos no nerviosos, mastocitos, pulmón |
La adenosina es el neuromodulador tranquilizante más importante del SNC. Su acción no es inhibidora en sentido clásico, sino moduladora y difusa: reduce la excitabilidad general del encéfalo. El receptor A1 es su principal diana, y lo hace hiperpolarizando las neuronas mediante apertura de canales GIRK (canales de K⁺ rectificadores hacia adentro acoplados a GPCR).
22.5.5. Adenosina y presión homeostática del sueño
La adenosina tiene un papel central en la regulación del sueño que conecta directamente con los circuitos GABAérgicos del ciclo sueño-vigilia vistos en el tema anterior.
Durante la vigilia, la actividad neuronal genera adenosina como producto del metabolismo energético (degradación de ATP). La adenosina se acumula progresivamente en el espacio extracelular del prosencéfalo basal y otras regiones encefálicas. Al activar los receptores A1 y A2A, inhibe los sistemas de arousal (neuronas colinérgicas del prosencéfalo basal, neuronas monoaminérgicas del tronco encefálico) y aumenta la actividad de las neuronas GABAérgicas del área preóptica que inducen el sueño. El resultado es una presión creciente hacia el sueño que se disipa durante este.
La presión homeostática del sueño es, en términos bioquímicos, acumulación de adenosina extracelular durante la vigilia. Cuanto más tiempo lleva despierta una persona, más adenosina se ha acumulado y mayor es la presión para dormir. El sueño aclarará esa adenosina, y la presión se disipará.
Cafeína: es el antagonista competitivo de los receptores de adenosina A1 y A2A. Al bloquearlos, impide el efecto tranquilizante y somnolífico de la adenosina acumulada, produciendo alerta y activación. La cafeína no elimina la adenosina ni la presión homeostática: esta sigue acumulándose. Cuando el efecto de la cafeína desaparece, la adenosina acumulada actúa de golpe, explicando la somnolencia de rebote.
La teofilina (broncodilatador) actúa por el mismo mecanismo, lo que explica sus efectos secundarios sobre el estado de ánimo y la irritabilidad.
Los circuitos GABAérgicos del área preóptica que median el inicio del sueño y sobre los que actúa la adenosina se describen en el apartado de funciones del sistema GABAérgico (Sinapsis GABAérgicas y glicinérgicas).
