23.1. Características generales de los NT no canónicos
Los neurotransmisores no canónicos se sitúan en sinapsis que no siguen el modelo clásico vesicular. Sus propiedades los distinguen radicalmente de los NT convencionales:
- No se almacenan en vesículas ni se liberan en cuantos.
- No actúan sobre receptores de membrana convencionales.
- Son liposolubles o gaseosos, lo que les permite atravesar libremente las membranas celulares por difusión.
- Actúan frecuentemente de forma retrógrada: son liberados por la postsinapsis y actúan sobre la presinapsis.
Los principales son gases (NO y CO) y lípidos (ácido araquidónico y endocannabinoides).
| NT | Tipo | Síntesis / enzima | Receptor / diana | Función principal | Relevancia clínica |
|---|---|---|---|---|---|
| NO | Gas radical libre | L-Arginina → NOS (BH4, FAD, FMN, NADPH) | Guanilato ciclasa soluble → ↑GMPc → PKG | Vasodilatación, LTP (mensajero retrógrado), antimicrobiano | Nitratos (angina), sildenafilo (PDE5i), desacoplamiento NOS en HTA |
| CO | Gas | Hemo → HO-1/HO-2 → CO + biliverdina + Fe²⁺ | Guanilato ciclasa soluble → ↑GMPc (baja afinidad) | Neuromodulación, vasodilatación, antiinflamatorio | Intoxicación: COHb → hipoxia tisular (O₂ 100%, cámara hiperbárica) |
| Anandamida (AEA) | Lípido (N-acil etanolamina) | NAPE → NAPE-PLD → AEA (activada por Ca²⁺) | CB1 (Gi) presináptico → ↓AMPc, ↓Ca²⁺, ↓liberación NT | Modulación retrógrada, analgesia, apetito, memoria | FAAH (diana terapéutica); THC activa CB1 con mayor potencia |
| 2-AG | Lípido (monoacilglicerol) | PIP2 → PLC → DAG → DAG lipasa → 2-AG (activada por mGluR1/5) | CB1 (Gi) presináptico (agonista pleno, más potente que AEA) | DSI, DSE, plasticidad sináptica a corto plazo | MAGL (diana terapéutica) |
| Ác. araquidónico | Lípido (ácido graso) | Fosfolípido → PLA2 → AA libre | Canales iónicos directamente; precursor de eicosanoides (COX, LOX) | Mediador inflamatorio, modulador de canales | AINEs (↓COX); aspirina (COX irreversible); celecoxib (COX-2 selectivo) |
23.2. Óxido nítrico (NO)
23.2.1. Síntesis de óxido nítrico
El NO es un gas incoloro e inodoro, radical libre, con una vida media muy corta (no más de 5 segundos). No tiene mecanismo de degradación enzimática específico: se inactiva espontáneamente por oxidación. Al ser un gas, difunde en todas direcciones sin restricción espacial.
L-Arginina + O₂ + NADPH → L-Citrulina + NO + NADP⁺
Enzima: óxido nítrico sintasa (NOS) (requiere FAD como cofactor)
La síntesis se activa cuando aumenta el Ca²⁺ intracelular: el Ca²⁺ se une a la calmodulina, y el complejo Ca²⁺-calmodulina activa la NOS.
Se han descrito tres isoformas de NOS con distribución y regulación distintas:
| Isoforma | Nombre | Regulación | Localización principal |
|---|---|---|---|
| NOS-I | NOSn (neural) | Constitutiva | Neuronas y otros tipos celulares |
| NOS-II | NOSi (inducible) | Inducible | Células inmunitarias; prácticamente todas las células |
| NOS-III | NOSe (endotelial) | Constitutiva | Endotelio vascular y músculo |
La mayoría de las células expresan al menos dos isoformas de NOS.
23.2.2. Mecanismo de acción del NO y sinapsis nitrérgica
La sinapsis nitrérgica invierte el sentido convencional de la transmisión. El potencial de acción en la neurona postsináptica provoca un aumento de Ca²⁺ que activa la NOS y genera NO. El NO difunde libremente hacia atrás, hasta la presinapsis, donde activa la guanilato ciclasa:
GTP → GMPc (guanilato ciclasa)
El aumento de GMPc en la presinapsis activa la proteín quinasa G (PKG), que fosforila proteínas efectoras y modula la liberación de neurotransmisor.
La sinapsis nitrérgica carece de vesículas, cuantos y receptores de membrana. El NO no es captado por un receptor: penetra directamente en la célula diana por difusión y activa enzimas intracelulares. Esta es la razón por la que se clasifica como NT no canónico.
23.2.3. Funciones del óxido nítrico
- Vasodilatación: el GMPc generado en el músculo liso vascular inhibe la miosina quinasa y cierra canales de Ca²⁺, impidiendo la contracción y produciendo relajación del vaso. Este es el mecanismo de acción de los nitratos (nitroglicerina/cafinitrina): donadores exógenos de NO que producen vasodilatación coronaria y periférica en el tratamiento de la angina de pecho.
- Memoria y LTP: el glutamato activa receptores NMDA en la postsinapsis, permitiendo la entrada de Ca²⁺ que activa la NOS postsináptica. El NO generado difunde retrogradamente a la presinapsis, donde aumenta el GMPc y potencia la liberación de más glutamato. Se establece así un circuito reverberante (engrama) que mantiene la actividad sináptica prolongada, base de la potenciación a largo plazo y de los procesos de memoria.
- Acción como radical libre: el NO se combina con el anión superóxido (O₂⁻) formando peroxinitrito, un oxidante muy reactivo que daña membranas, fragmenta el ADN y puede atacar la mitocondria. Si alcanza la mitocondria, libera citocromo C y activa la cascada de las caspasas, desencadenando la apoptosis.
- Función antimicrobiana: la NOSi inducible en células inmunitarias produce grandes cantidades de NO que destruyen microorganismos patógenos por daño oxidativo.
- Efecto broncodilatador: el NO producido por el endotelio pulmonar y el músculo liso bronquial relaja la musculatura bronquial; se estudia su papel en el asma.
El sildenafilo (Viagra) no es un donador de NO: actúa bloqueando la fosfodiesterasa-5 (PDE5), la enzima que degrada el GMPc en el músculo liso del cuerpo cavernoso. Al impedir la degradación del GMPc, prolonga la vasodilatación mediada por NO y facilita la erección. El mismo mecanismo explica su uso en hipertensión pulmonar (sildenafilo/tadalafilo).
23.2.4. Óxido nítrico y patología
NO "bueno" vs. NO "malo": el NO producido por las NOS constitutivas (NOS-I y NOS-III) en condiciones fisiológicas tiene efectos protectores: vasodilatación, modulación sináptica, defensa antimicrobiana controlada. El NO producido en exceso por la NOSi en procesos inflamatorios crónicos o patológicos puede contribuir a daño tisular, apoptosis neuronal y progresión tumoral. La distinción entre ambos depende de la cantidad, el contexto celular y la presencia de superóxido.
En la enfermedad de Parkinson, la muerte de neuronas dopaminérgicas de la sustancia negra libera NO patológico que amplifica la apoptosis en los circuitos de los ganglios basales.
En cuanto a la metástasis, las células endoteliales sanas producen NO de forma continua, lo que impide la adhesión de células tumorales circulantes a la pared vascular. La disfunción endotelial reduce este efecto protector y facilita la extravasación tumoral.
23.3. Monóxido de carbono (CO)
El monóxido de carbono es el segundo gas neuroactivo caracterizado en el SNC. Comparte con el NO el mecanismo de acción central (activación de la guanilato ciclasa → ↑GMPc), pero es mucho menos potente y tiene una vida media más larga.
CO como neuromodulador: el CO producido endógenamente en concentraciones fisiológicas actúa como mensajero gaseoso que activa la guanilato ciclasa soluble, eleva el GMPc y produce efectos neuromoduladores, vasodilatadores y antiinflamatorios. Es distinto del CO exógeno de la intoxicación, que actúa por un mecanismo de hipoxia tisular.
23.3.1. Síntesis del CO
El CO endógeno se produce como subproducto de la degradación del grupo hemo:
Síntesis del CO:
Hemo + O₂ + NADPH → Biliverdina + Fe²⁺ + CO
Enzima: hemo oxigenasa (HO)
Existen dos isoformas relevantes:
- HO-1 (inducible): se expresa ante estrés oxidativo, hipoxia, lipopolisacáridos bacterianos y metales pesados. Es parte de la respuesta celular al daño.
- HO-2 (constitutiva): expresión basal en neuronas del cerebelo, hipocampo, neuronas del plexo mientérico y endotelio. Genera el CO con función neuromoduladora fisiológica.
La biliverdina generada en la misma reacción se convierte en bilirrubina por la biliverdina reductasa. La bilirrubina es un potente antioxidante endógeno. La vía HO no es solo una fuente de CO: es un sistema integrado de defensa frente al estrés oxidativo que genera tres productos activos (CO, biliverdina y Fe²⁺ liberador de ferritina).
23.3.2. Mecanismo de acción y funciones del CO
El CO activa la guanilato ciclasa soluble (sGC) uniéndose a su grupo hemo, al igual que el NO, aunque con afinidad mucho menor. El aumento de GMPc resultante activa la PKG y produce efectos análogos a los del NO pero de menor magnitud.
Sus funciones fisiológicas incluyen neuromodulación en el hipocampo y el cerebelo, vasodilatación (especialmente en la circulación esplácnica y pulmonar), efecto antiinflamatorio (inhibe la producción de citocinas proinflamatorias por la microglía) y regulación del ciclo circadiano en el núcleo supraquiasmático.
Intoxicación por CO: el CO exógeno (combustión incompleta de carburantes, incendios) actúa por un mecanismo completamente distinto al CO endógeno. Su afinidad por la hemoglobina es unas 240 veces mayor que la del O₂, formando carboxihemoglobina (COHb), que no transporta O₂. Además inhibe la cadena respiratoria mitocondrial uniéndose al citocromo a₃. El resultado es hipoxia tisular grave con daño neuronal preferente. El tratamiento es O₂ al 100% (desplaza competitivamente el CO de la hemoglobina) o cámara hiperbárica en casos graves.
La intoxicación por CO y la función neuromoduladora del CO endógeno son mecanismos completamente distintos. El CO endógeno actúa a concentraciones nanomolares activando la guanilato ciclasa. El CO exógeno actúa a concentraciones micromolares bloqueando el transporte de O₂ y la cadena respiratoria. Misma molécula, mecanismos y consecuencias opuestos según la concentración y la fuente.
23.4. Endocannabinoides y ácido araquidónico
Los mediadores lipídicos forman la familia más reciente de neurotransmisores no canónicos. A diferencia de los gases, son moléculas lipídicas de cadena larga que se sintetizan a demanda en la membrana postsináptica y actúan retrógradamente sobre receptores específicos en la presinapsis.
23.4.1. Ácido araquidónico como mediador sináptico
El ácido araquidónico (AA) es un ácido graso poliinsaturado de 20 carbonos (omega-6) que forma parte de los fosfolípidos de la membrana celular en posición sn-2. No se almacena libre: se libera a demanda cuando la membrana es activada.
Liberación del ácido araquidónico:
Fosfolípido de membrana → AA libre + lisofosfoglicérido
Enzima: fosfolipasa A2 (PLA2), activada por ↑Ca²⁺ intracelular o por activación de receptores acoplados a Gq
Una vez libre, el AA puede actuar como mensajero lipídico directo (modula canales iónicos y enzimas) o ser metabolizado por tres vías enzimáticas:
- Vía COX(ciclooxigenasa): → prostaglandinas y tromboxanos.
- Vía LOX(lipoxigenasa): → leucotrienos.
- Vía CYP450(citocromo P450): → EET (ácidos epoxieicosatrienoicos, vasoactivos)
Los AINEs (antiinflamatorios no esteroideos: ibuprofeno, naproxeno, diclofenaco) inhiben la COX-1 y COX-2, bloqueando la síntesis de prostaglandinas y tromboxanos a partir del ácido araquidónico. La aspirina inhibe la COX de forma irreversible (acetilación); el resto de AINEs la inhiben de forma reversible. Los inhibidores selectivos de COX-2 (celecoxib) fueron diseñados para reducir los efectos gastrointestinales de la inhibición de COX-1.
La farmacología completa de los eicosanoides, las prostaglandinas y los AINEs se estudia en Farmacología y en Patología General. En Fisiología General interesa el ácido araquidónico como mensajero lipídico sináptico y como precursor de mediadores inflamatorios cuya síntesis puede ser bloqueada farmacológicamente.
23.4.2. Endocannabinoides: anandamida y 2-AG
Los endocannabinoides son los ligandos endógenos de los receptores cannabinoides, los mismos receptores activados por el THC del cannabis. Los dos principales son la anandamida (AEA, araquidonoiletanolamida) y el 2-araquidonoilglicerol (2-AG), ambos derivados del ácido araquidónico.
Endocannabinoides: lípidos bioactivos derivados del ácido araquidónico que actúan como mensajeros sinápticos retrógrados. Se sintetizan a demanda en la membrana postsináptica, difunden hacia la presinapsis y reducen la liberación de neurotransmisor activando receptores CB1 presinápticos acoplados a Gi.
23.4.3. Síntesis y degradación
La síntesis de anandamida y 2-AG sigue rutas distintas porque parten de precursores de membrana distintos, pero ambas comparten el disparador, la activación intensa de la postsinapsis.
- La anandamida responde principalmente a la entrada de Ca²⁺.
- El 2-AG responde a la activación de receptores metabotrópicos acoplados a Gq.
Esta diferencia es la responsable de que los dos endocannabinoides se liberen en condiciones ligeramente distintas y pueden tener pesos relativos diferentes según el tipo de sinapsis y el patrón de activación.
El 2-AG es cuantitativamente el endocannabinoide más abundante en el SNC y se considera el agonista endógeno principal de CB1, mientras que la anandamida actúa a concentraciones más bajas pero con mayor afinidad por CB1 que el 2-AG.
Síntesis de endocannabinoides (postsinapsis):
Anandamida (AEA): Fosfatidiletanolamina de membrana → N-araquidonoil-fosfatidiletanolamina → AEA
Enzima final: NAPE-PLD (fosfolipasa D específica), activada por: ↑Ca²⁺ postsináptico.
2-Araquidonoilglicerol (2-AG): Fosfatidil-inositol → DAG (diacilglicerol) → 2-AG
Enzimas: PLC (genera DAG) → DAG lipasa (genera 2-AG)
Activada por: activación de receptores Gq postsinápticos (mGluR1/5, receptores muscarínicos)
Degradación:
- AEA: hidrolizada por FAAH (fatty acid amide hydrolase)en la membrana postsináptica o glial → ácido araquidónico + etanolamina.
- 2-AG: hidrolizado por MAGL (monoacilglicerol lipasa) en la presinapsis → ácido araquidónico + glicerol.
Los endocannabinoides no se almacenan: se sintetizan en segundos cuando la postsinapsis se activa intensamente (↑Ca²⁺ o activación de mGluR) y se degradan con rapidez. Su señal es transitoria y localizada: solo actúan sobre la presinapsis inmediatamente adyacente a donde se sintetizan.
23.4.4. Receptores cannabinoides
Los receptores cannabinoides se descubrieron en 1988 buscando la diana del THC, antes de que se conociera la existencia de ligandos endógenos.
El hallazgo de que el cerebro tenía receptores específicos para un componente del cannabis llevó directamente a la búsqueda de los ligandos endógenos que los activaban: la anandamida se identificó en 1992 y el 2-AG en 1995.
De los dos receptores clonados, CB1 es el más relevante en el SNC y uno de los GPCRs más abundantes del encéfalo. El CB2 predomina en el sistema inmune y tiene expresión neuronal mucho más limitada, aunque su papel en la microglía y la neuroinflamación ha ganado interés clínico en los últimos años.
| Receptor | Proteína G | Localización | Función principal |
|---|---|---|---|
| CB1 | Gi/Go → ↓AMPc, cierre Ca²⁺, apertura K⁺ | Presinapsis neuronal (SNC: corteza, hipocampo, cerebelo, ganglios basales) | Reducción de la liberación de NT; modulación de LTP y LTD; control del dolor, apetito, memoria |
| CB2 | Gi → ↓AMPc | Sistema inmune (microglía, linfocitos, bazo); escasa expresión neuronal | Modulación de la respuesta inflamatoria e inmune; neuroprotección |
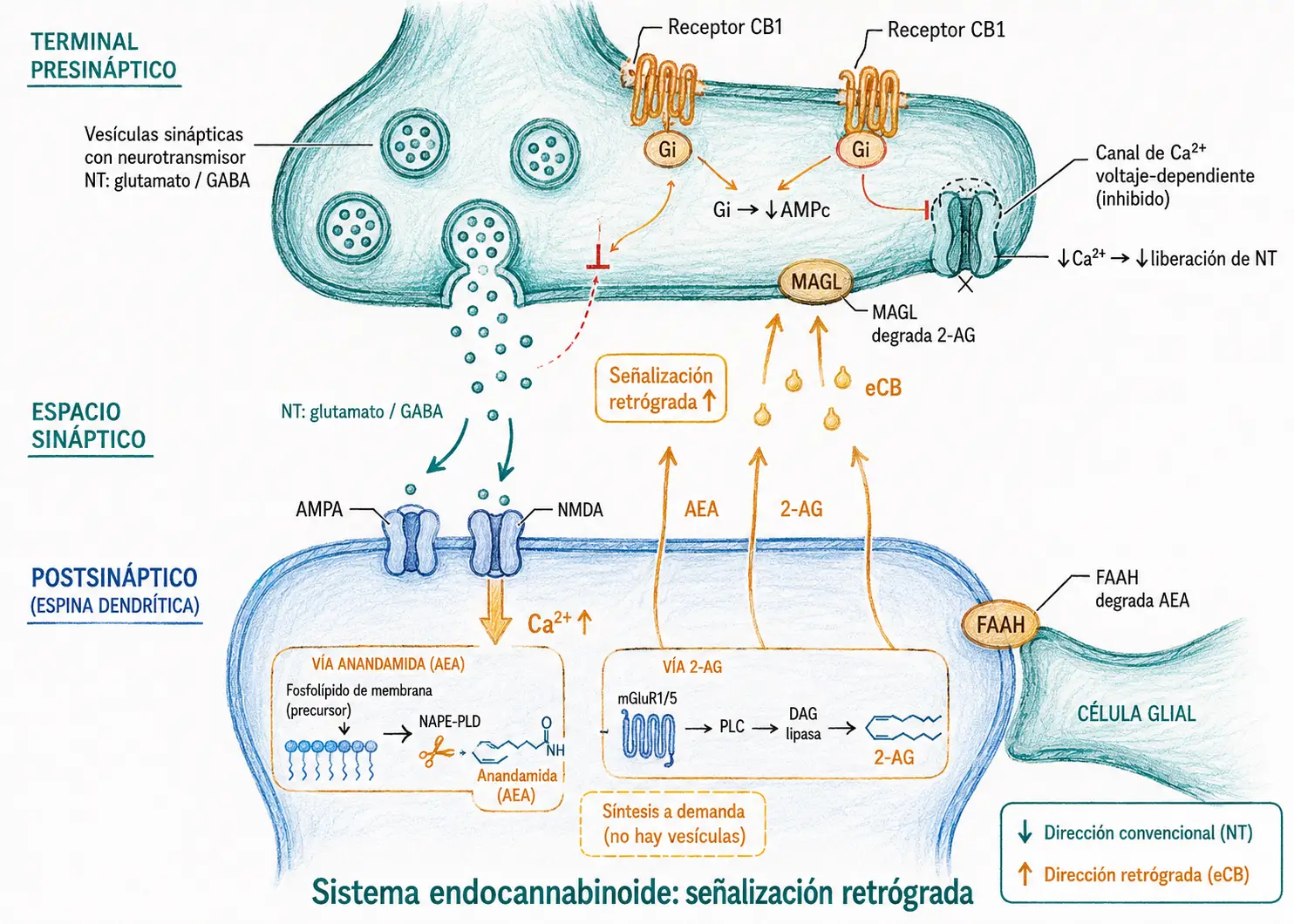
23.4.5. Mecanismo de la señalización retrógrada endocannabinoide
La señalización retrógrada endocannabinoide resuelve un problema concreto de la fisiología sináptica: ¿cómo sabe la presinapsis que la postsinapsis ya está saturada?
Los neurotransmisores clásicos solo viajan en una dirección, por lo que la presinapsis libera de forma ciega, sin información sobre el estado de activación de la célula que está estimulando. Los endocannabinoides invierten ese flujo de información: cuando la postsinapsis se activa en exceso, fabrica la señal de freno y la envía hacia atrás. La presinapsis recibe el mensaje y reduce la liberación. Es un sistema de control local, rápido y sin intermediarios.
Secuencia de la señalización retrógrada endocannabinoide:
- Estimulación intensa de la neurona postsináptica → ↑Ca²⁺ intracelular postsináptico (o activación de mGluR1/5).
- La postsinapsis sintetiza endocannabinoides (AEA o 2-AG) a demanda.
- Los endocannabinoides difunden retrógradamente a través de la hendidura sináptica hasta la membrana de la terminal presináptica.
- Se unen a receptores CB1 presinápticos → Gi/Go → ↓AMPc + cierre de canales Ca²⁺ dependientes de voltaje.
- La reducción de Ca²⁺ en la presinapsis reduce la liberación de neurotransmisor (glutamato, GABA u otros).
- El efecto se denomina DSI (Depolarization-induced Suppression of Inhibition, si el NT inhibido es GABA) o DSE (Depolarization-induced Suppression of Excitation, si el NT inhibido es glutamato).
- Los endocannabinoides son degradados por FAAH o MAGL, terminando la señal.
El sistema endocannabinoide es una forma de retroalimentación negativa sináptica: cuando la neurona postsináptica se activa en exceso, sintetiza endocannabinoides que reducen la liberación del neurotransmisor que la estaba activando. Es un mecanismo de autoprotección contra la sobreestimulación, relevante tanto en la plasticidad sináptica como en la neuroprotección frente a la excitotoxicidad.
La magnitud y duración de esta supresión depende de la cantidad de endocannabinoide producido y de la velocidad a la que FAAH y MAGL lo degradan.
En condiciones normales la señal dura segundos, lo suficiente para amortiguar un pico de actividad excesiva. Cuando FAAH o MAGL están inhibidos (farmacológicamente o por déficit genético), los endocannabinoides se acumulan y la supresión se prolonga. Esta es la razón por la que los inhibidores de FAAH se investigan como analgésicos y ansiolíticos. No introducen un ligando exógeno, sino que prolongan la señal endógena ya generada por el propio organismo.
23.4.6. Funciones del sistema endocannabinoide
El sistema endocannabinoide interviene en la regulación de múltiples funciones fisiológicas. Su distribución en el SNC explica las regiones más afectadas por el cannabis:
- Control del dolor: los receptores CB1 en el asta posterior medular y en el cerebro reducen la transmisión nociceptiva, un efecto que el cannabis explota pero que existe fisiológicamente.
- Regulación del apetito: los receptores CB1 en el hipotálamo lateral y el núcleo arcuato estimulan el apetito. El rimonabant, un antagonista CB1, fue aprobado como antiobesidad pero retirado por efectos psiquiátricos graves.
- Memoria y aprendizaje: la activación de CB1 en el hipocampo facilita el olvido de memorias aversivas (extinción del miedo condicionado). Esto tiene aplicaciones terapéuticas potenciales en el TEPT.
- Coordinación motora: los receptores CB1 en el cerebelo y los ganglios basales modulan la precisión y la fluidez del movimiento. La ataxia por cannabis es una manifestación directa de la activación de CB1 en el cerebelo.
La distribución regional de los receptores CB1 en el SNC no es uniforme. Esa distribución explica directamente los efectos del cannabis.
Las regiones con mayor densidad de CB1 son el hipocampo (memoria), el cerebelo y los ganglios basales (coordinación motora), la corteza prefrontal (funciones ejecutivas y percepción temporal) y el hipotálamo lateral (apetito).
El sistema límbico, especialmente la amígdala, tiene también alta densidad de CB1, lo que explica los efectos del cannabis sobre la ansiedad y el procesamiento emocional.
Las regiones del tronco encefálico que controlan la respiración y la frecuencia cardíaca tienen densidad de CB1 muy baja. Esto explica por qué no existe una dosis letal de cannabis por depresión respiratoria, a diferencia de los opioides o los barbitúricos.
Cannabis y THC: el Δ9-tetrahidrocannabinol (THC) activa los receptores CB1 con mayor potencia y durante más tiempo que los endocannabinoides endógenos (que son degradados en segundos por FAAH/MAGL). Esta activación sostenida y no fisiológica produce los efectos del cannabis: euforia (sistema de recompensa mesolímbico), alteración de la memoria a corto plazo (hipocampo), analgesia (médula), aumento del apetito (hipotálamo) y ataxia (cerebelo). El uso crónico produce down-regulation de CB1, tolerancia y, en individuos predispuestos, aumento del riesgo de psicosis.
El cannabidiol (CBD) no activa directamente los receptores CB1 ni CB2 con afinidad relevante. Sus mecanismos son múltiples y menos caracterizados: inhibición de la FAAH (aumenta niveles de anandamida endógena), antagonismo de receptores GPR55, activación de receptores TRPV1 y modulación de canales de Na⁺. Sus efectos ansiolíticos y antiepilépticos (epidiolex está aprobado para algunas epilepsias refractarias pediátricas) no dependen de la activación directa de CB1.
