21.1. Generalidades de las sinapsis GABAérgicas y glicinérgicas
La mayoría de los neurotransmisores del SNC son aminoácidos o derivados de aminoácidos. El GABA (ácido γ-aminobutírico) y la glicina son los dos principales neurotransmisores inhibidores del SNC, y su función es contrarrestar la excitación glutamatérgica para mantener el equilibrio excitación/inhibición del encéfalo y la médula espinal.
El GABA actúa como inhibidor en prácticamente todas las regiones encefálicas. En la corteza cerebral, aproximadamente el 20% de las neuronas son GABAérgicas y forman circuitos locales inhibitorios sin los cuales la actividad cortical se volvería paroxística.
La glicina tiene una distribución más restringida. Su acción como neurotransmisor se concentra en el tronco encefálico y la médula espinal: más del 50% de las sinapsis inhibidoras medulares utilizan glicina como neurotransmisor. Las interneuronas glicinérgicas inhiben las motoneuronas del músculo antagonista durante el movimiento, permitiendo la coordinación motora fluida.
Tanto el GABA como la glicina inhiben la generación de potenciales de acción aumentando la conductancia al Cl⁻ y acercando el potencial de membrana al potencial de equilibrio del cloro. El bloqueo experimental de sus receptores (estricnina para glicina, bicuculina para GABA-A) produce convulsiones y espasticidad, lo que ilustra que la inhibición es activa y tan necesaria como la excitación para el funcionamiento normal del SNC.
21.2. Síntesis y eliminación del GABA
El GABA se sintetiza a partir del glutamato en un único paso enzimático:
Glutamato → GABA
Enzima: ácido glutámico descarboxilasa (GAD)
Cofactor: fosfato de piridoxal (derivado de la vitamina B6)
La GAD se expresa exclusivamente en neuronas GABAérgicas.
La GAD se expresa exclusivamente en neuronas GABAérgicas y es el marcador bioquímico específico de este tipo neuronal. Existen dos isoformas: GAD65 (asociada a la membrana vesicular, responsable del GABA liberado en la sinapsis) y GAD67 (citosólica, responsable del pool metabólico basal).[/mnv_caja]
Convulsiones por déficit de vitamina B6: la GAD depende del fosfato de piridoxal como cofactor. La deficiencia de piridoxina reduce la síntesis de GABA y puede provocar convulsiones neonatales. Este mecanismo se identificó tras la muerte de neonatos alimentados con una fórmula láctea industrialmente deficiente en vitamina B6. Las convulsiones dependientes de piridoxina son hoy una causa reconocida de epilepsia neonatal tratable con suplementación.
La inactivación del GABA en la hendidura sináptica se produce principalmente por recaptación mediante transportadores específicos de la familia GAT (GABA Transporters). El subtipo más importante es el GAT-1, expresado en terminales presinápticas y astrocitos, que cotransporta GABA con Na⁺ y Cl⁻ en favor del gradiente de sodio.
Una vez recaptado, el GABA se metaboliza en dos pasos enzimáticos hacia succinato, que entra en el ciclo de Krebs:
Degradación del GABA:
- GABA → Succínico semialdehído: GABA aminotransferasa (GABA-T), requiere fosfato de piridoxal.
- Succínico semialdehído → Succinato: succínico semialdehído deshidrogenasa (SSADH).
El succinato resultante entra en el ciclo de Krebs como intermediario. Esta vía se denomina «shunt del GABA» y representa una conexión entre el metabolismo del glutamato y el ciclo de Krebs.
Tiagabina: este antiepiléptico inhibe selectivamente GAT-1, impidiendo la recaptación del GABA y prolongando su efecto inhibidor en la hendidura sináptica. Es un ejemplo de que los transportadores de recaptación son dianas farmacológicas directas, igual que los EAAT en el sistema glutamatérgico (→ T20: Sinapsis glutamatérgicas).
Valproato y vigabatrina: el valproato sódico inhibe la GABA aminotransferasa (GABA-T), reduciendo la degradación de GABA y elevando sus niveles sinápticos. La vigabatrina (γ-vinil-GABA) es un inhibidor irreversible de GABA-T con el mismo mecanismo. Ambos son antiepilépticos de uso clínico cuyo mecanismo molecular ancla directamente en la vía de degradación del GABA.
21.3. Síntesis y eliminación de la glicina
La glicina se sintetiza principalmente a partir de la serina:
Síntesis de glicina:
Serina → Glicina
Enzima: serina hidroximetiltransferasa (SHMT)
Cofactor: tetrahidrofolato (THF), que acepta el grupo metileno transferido
La SHMT es reversible y cataliza también la reacción inversa (glicina + formaldehído → serina).
La degradación de la glicina se produce principalmente a través del sistema de escisión de la glicina (GCS, glycine cleavage system), un complejo multienzimático mitocondrial formado por cuatro proteínas (P, H, T y L). El GCS convierte la glicina en CO₂, NH₄⁺ y un fragmento de un carbono que se transfiere al tetrahidrofolato. Este sistema regula los niveles intracelulares de glicina libre y es su principal vía de catabolismo.
La inactivación sináptica de la glicina se produce por recaptación mediante transportadores GlyT1 (astrocitario) y GlyT2 (neuronal presináptico). GlyT1 también regula los niveles de glicina disponibles para el sitio coagonista del receptor NMDA (ver en el Tema 20 · Sinapsis glutamatérgicas).
Hiperglicinemia no cetósica: enfermedad metabólica hereditaria grave causada por mutaciones en los genes que codifican las proteínas del sistema de escisión de la glicina (GCS), principalmente en la proteína P (gen GLDC). La incapacidad para degradar la glicina produce su acumulación en plasma, orina y LCR. Los neonatos presentan somnolencia, hipotonía, mioclonías y apneas; los supervivientes desarrollan retraso mental severo. El tratamiento combina la restricción de glicina en la dieta con el antagonismo del receptor NMDA (para bloquear el exceso de estimulación del sitio coagonista).
Un error frecuente es atribuir la hiperglicinemia no cetósica a «alteraciones en los transportadores de glicina». La causa es la disfunción del sistema de escisión de la glicina (GCS): los transportadores GlyT son dianas terapéuticas en investigación, no la causa de la enfermedad.
21.4. Receptores GABA
Existen tres categorías de receptores GABAérgicos con propiedades moleculares y funcionales bien diferenciadas:
| Receptor | Tipo | Ion | Localización | Relevancia farmacológica |
|---|---|---|---|---|
| GABA-A | Ionotrópico (canal Cl⁻) | Cl⁻ entra | Corteza, hipocampo, cerebelo, sistema límbico | BZD, barbitúricos, alcohol, anestésicos, antiepilépticos |
| GABA-B | Metabotrópico (prot. Gi) | K⁺ sale (↑ conductancia K⁺) / ↓ Ca²⁺ presináptico | Amplia distribución, pre y postsináptico | Baclofeno (relajante muscular, espasticidad) |
| GABA-A-ρ | Ionotrópico (canal Cl⁻) | Cl⁻ entra | Predominantemente retina | Insensible a BZD y barbitúricos; subunidades ρ |
21.4.1. Receptor GABA-A: estructura y sitios farmacológicos
El receptor GABA-A es el principal receptor ionotrópico inhibidor del SNC y la diana farmacológica más importante de este tema. Es un canal pentamérico de Cl⁻. Al unirse el GABA, se abre y entra Cl⁻, hiperpolarizando la membrana y generando un potencial postsináptico inhibidor (PPSI).
La composición más frecuente en el SNC adulto es 2α + 2β + 1γ, pero existen al menos 19 subunidades distintas (α1-6, β1-3, γ1-3, δ, ε, θ, π y ρ), y la combinación específica de subunidades determina las propiedades biofísicas del canal, su distribución regional y su sensibilidad a los distintos fármacos.
Los sitios de unión farmacológica están físicamente separados dentro del receptor:
- El GABA se une en la interfaz entre subunidades α y β, en dos puntos simétricos del pentámero.
- Las benzodiazepinas se unen en la interfaz α/γ: por eso solo potencian receptores que contienen la subunidad γ; sin ella (como los receptores con subunidad δ), las benzodiazepinas clásicas no tienen efecto.
- Los barbitúricos se unen dentro del poro del canal (sitio intracanal en subunidades β) y pueden mantenerlo abierto más tiempo, incluso en ausencia de GABA a dosis altas.
- Los neuroesteroides endógenos (como la alopregnanolona) actúan sobre receptores que contienen subunidad δ, predominantemente extrasinápticos, que median la llamada inhibición tónica (conductancia GABAérgica de baja intensidad y continua que regula el umbral de excitabilidad global).
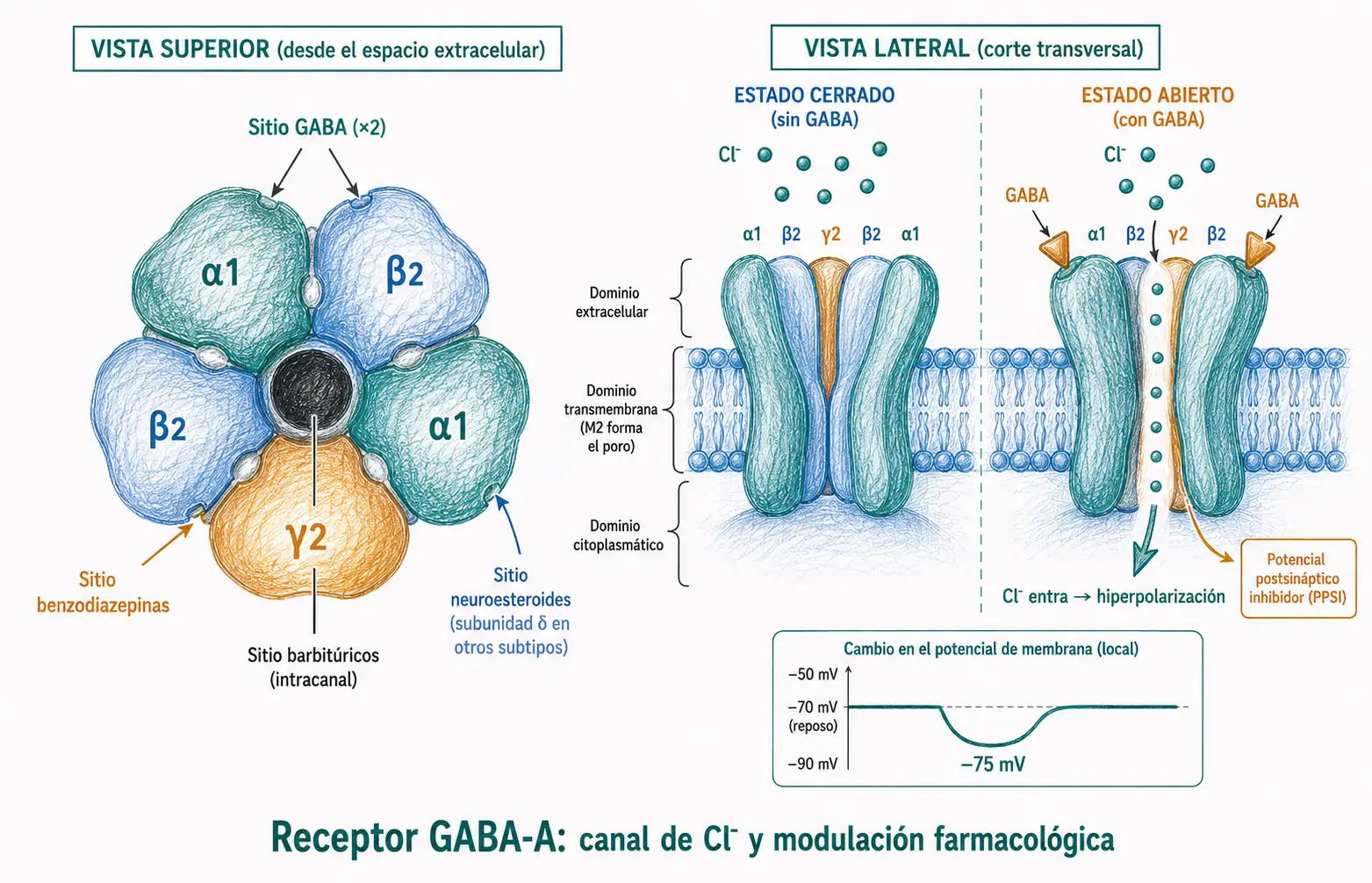
La nomenclatura histórica separaba los receptores formados por subunidades ρ como «GABA-C». La IUPHAR los reclasificó en 2008 como GABA-A-ρ: son receptores GABA-A con propiedades específicas (cinética más lenta, insensibilidad a benzodiazepinas y barbitúricos) por la composición exclusiva de subunidades ρ. En libros más antiguos puede aparecer la denominación GABA-C, que debe considerarse obsoleta.
21.4.2. Receptor GABA-B
El receptor GABA-B es metabotrópico, acoplado a proteína G<sub>i/o</sub>. Actúa a través de dos mecanismos: postsináptico, abre canales de K⁺ (hiperpolarización lenta y prolongada); presináptico, como autorreceptor, reduce la entrada de Ca²⁺ en la terminal presináptica y disminuye la liberación de GABA (y en sinapsis heterólogas, de otros neurotransmisores).
Baclofeno: agonista selectivo del receptor GABA-B, utilizado para reducir la espasticidad muscular en esclerosis múltiple y lesiones medulares. Al activar GABA-B en las motoneuronas y en las interneuronas de la médula espinal, reduce el tono muscular aumentado por la pérdida de control inhibidor descendente.
21.5. Receptores de glicina
Los receptores de glicina son ionotrópicos, pentaméricos y funcionalmente análogos al GABA-A: al abrirse, aumentan la conductancia al Cl⁻ e hipopolarizan la membrana postsináptica, generando PPSI.
La composición más frecuente es 3α + 2β. Cada subunidad α tiene un sitio de unión para la glicina, lo que significa que el receptor requiere la unión de hasta tres moléculas de glicina para su activación completa, aunque puede abrirse parcialmente con menos.
La estricnina es el antagonista competitivo del receptor de glicina (se une al mismo sitio que la glicina en la subunidad α y bloquea el canal). Produce convulsiones, espasmos musculares dolorosos y muerte por parálisis respiratoria. Es el veneno clásico de los libros de farmacología y el reactivo de laboratorio estándar para bloquear la inhibición glicinérgica experimentalmente. Distinguirla de la bicuculina (antagonista GABA-A) es una pregunta frecuente.
La hiperaplexia (enfermedad del sobresalto) es una enfermedad hereditaria causada por mutaciones en las subunidades α del receptor de glicina (gen GLRA1) que impiden la unión o la apertura normal del canal. Los pacientes presentan hipertonía muscular neonatal y respuestas exageradas, bruscas e incontrolables ante estímulos auditivos o táctiles inesperados (por falta de inhibición glicinérgica en el tronco encefálico y la médula). El tratamiento de elección es el clonazepam (benzodiazepina), que compensa la pérdida de inhibición activando los receptores GABA-A.
21.6. GABA excitador durante el desarrollo: el switch GABAérgico
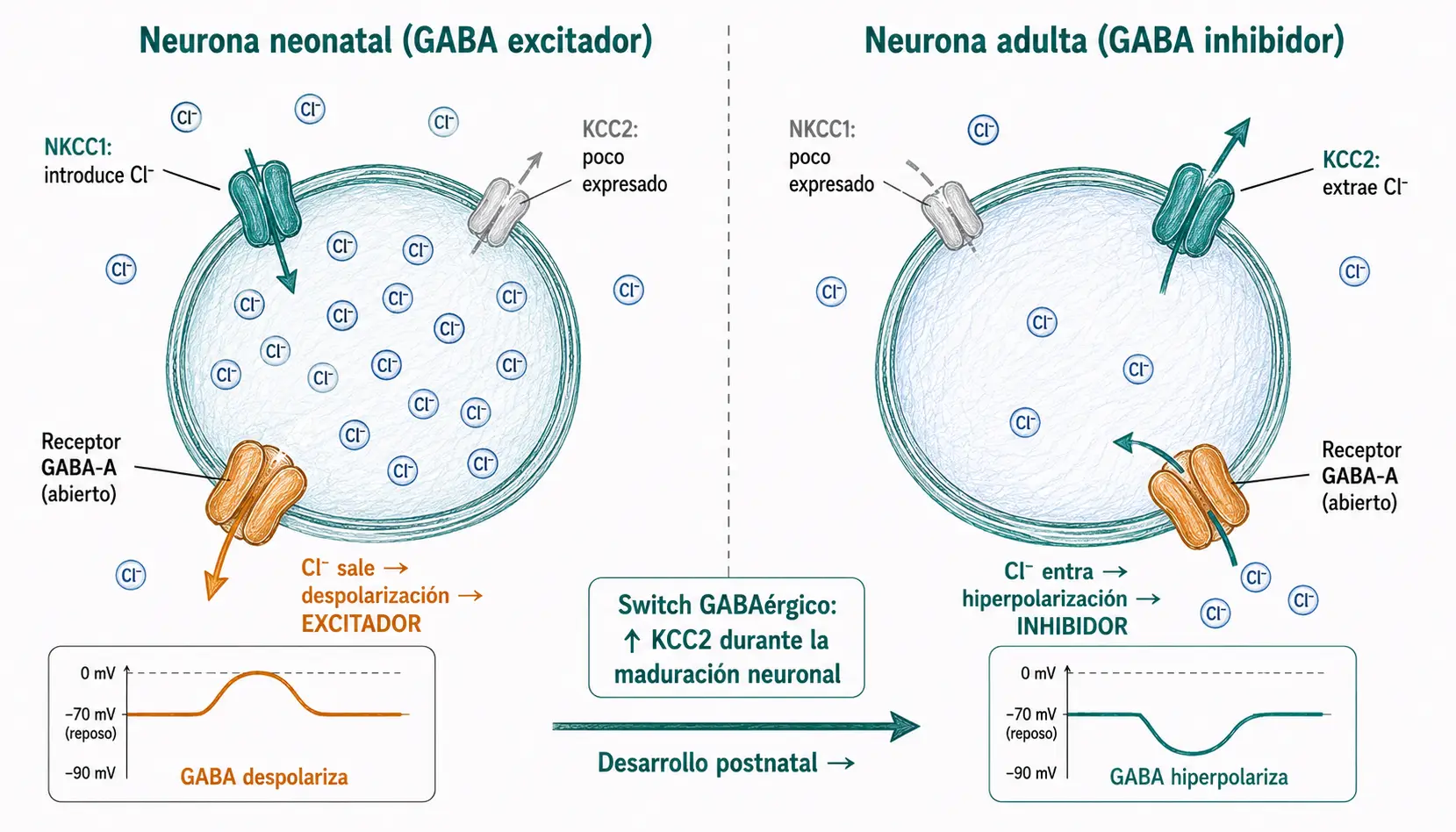
Un hecho que sorprende al estudiante es que el GABA, siendo el principal inhibidor del SNC adulto, actúa como excitador durante el desarrollo embrionario y neonatal temprano.
Esto se debe a que la dirección del flujo de Cl⁻ a través del canal GABA-A depende del gradiente electroquímico del cloro, que está determinado por el potencial de equilibrio del Cl⁻ (ECl). En neuronas inmaduras, la concentración intracelular de Cl⁻ es alta porque el cotransportador NKCC1 (que introduce Cl⁻ en la célula) predomina sobre KCC2 (que extrae Cl⁻). Por eso el ECl está más positivo que el potencial de membrana: cuando el canal se abre, el Cl⁻ sale en lugar de entrar, despolarizando la célula.
Durante la maduración neuronal, la expresión de KCC2 aumenta progresivamente y la de NKCC1 disminuye. Cuando KCC2 predomina, el Cl⁻ intracelular baja, ECl se hace más negativo que el potencial de membrana, y a partir de ese momento el GABA pasa a ser inhibidor. Este cambio se denomina switch GABAérgico.
21.7. Farmacología del sistema GABAérgico
El receptor GABA-A posee múltiples sitios de unión para sustancias distintas del GABA, lo que lo convierte en una de las dianas farmacológicas más importantes del SNC.
El receptor GABA-A es una de las dianas farmacológicas más importantes del SNC. Todos los fármacos que actúan sobre él son moduladores alostéricos positivos: potencian el efecto del GABA sobre el canal sin activarlo directamente (excepto los barbitúricos a dosis altas).
Las benzodiazepinas (diazepam, lorazepam, clonazepam, midazolam) se unen a la interfaz α/γ y aumentan la frecuencia de apertura del canal ante el GABA. No abren el canal en ausencia de GABA. Producen ansiolisis, relajación muscular, sedación y efecto anticonvulsivante. Solo actúan sobre receptores que contienen subunidad γ.
Los barbitúricos (fenobarbital, tiopental) se unen al poro intracanal y prolongan el tiempo de apertura del canal. A dosis anestésicas pueden activar el canal incluso sin GABA. Su margen terapéutico es más estrecho que el de las benzodiazepinas: a dosis altas producen depresión respiratoria grave.
El etanol potencia la transmisión GABAérgica actuando preferentemente sobre receptores con subunidad δ (extrasinápticos, de inhibición tónica) a concentraciones bajas y fisiológicamente relevantes. A concentraciones más altas actúa también sobre los receptores sinápticos. Además, el etanol inhibe los receptores NMDA de glutamato: esta combinación de potenciación GABAérgica e inhibición glutamatérgica explica su efecto depresor generalizado del SNC.
Las benzodiazepinas, los barbitúricos y el etanol actúan sobre sitios distintos del receptor GABA-A pero todos potencian la inhibición GABAérgica. Cuando se combinan, sus efectos son supraditivos: la presencia de un fármaco unido al receptor amplifica el efecto del siguiente. Esta es la base del riesgo letal de combinar alcohol con benzodiazepinas o barbitúricos: la depresión respiratoria puede ser fatal a dosis que individualmente serían seguras.
La bicuculina es el antagonista competitivo del sitio GABA en el receptor GABA-A (bloquea la interfaz α/β). La picrotoxina es un bloqueante de canal (antagonista no competitivo): ocupa físicamente el poro e impide el paso del Cl⁻ independientemente de si el GABA está unido. Ambas producen convulsiones y se usan como herramientas de investigación. Distinguir su mecanismo (competitivo vs. no competitivo) es un objetivo clásico de examen.
21.8. Funciones del sistema GABAérgico e inhibidor
El GABA y la glicina tienen funciones que van más allá de la simple oposición a la excitación.
- Regulación del umbral de excitabilidad cortical. Las interneuronas GABAérgicas corticales fijan el umbral a partir del cual una neurona piramidal puede disparar. Sin esta inhibición de fondo, cualquier excitación se propagaría de forma incontrolada. Las epilepsias son el ejemplo clínico más directo de fallo de este mecanismo.
- Control del ciclo sueño-vigilia. Las neuronas del área preóptica ventrolateral del hipotálamo son GABAérgicas e inhiben los centros de arousal (locus coeruleus, núcleo del rafe, hipotálamo posterior) durante el sueño NREM. Los fármacos hipnóticos (benzodiazepinas, zolpidem) explotan este circuito para inducir el sueño.
- Regulación de la ansiedad. El sistema límbico contiene alta densidad de receptores GABA-A. La reducción de la actividad GABAérgica en la amígdala se asocia a respuestas de ansiedad exageradas: este es el mecanismo por el que las benzodiazepinas son ansiolíticos eficaces.
- Coordinación motora y tono muscular. Las interneuronas de Purkinje del cerebelo son GABAérgicas y regulan la precisión del movimiento. Las interneuronas glicinérgicas medulares inhiben las motoneuronas del músculo antagonista, coordinando la contracción agonista-antagonista (reflejo de inhibición recíproca de Sherrington).
Inhibición shunting. Cuando el potencial de equilibrio del Cl⁻ está cerca del potencial de reposo (como ocurre en las dendritas de muchas neuronas maduras), la apertura de canales GABA-A no produce hiperpolarización apreciable, pero sí aumenta la conductancia de membrana. Al «cortocircuitar» la membrana, cualquier corriente excitadora que intente despolarizar esa zona debe generar una diferencia de potencial mayor para el mismo efecto. Esta inhibición por aumento de conductancia, sin cambio de potencial, se denomina inhibición shunting y es un mecanismo inhibidor tan eficaz como la hiperpolarización clásica.
21.9. Sistema GABAérgico y adicción
Las drogas de abuso producen sus efectos de recompensa en la intersección entre el sistema GABAérgico y el dopaminérgico.
Las neuronas dopaminérgicas de la vía mesolímbica (área tegmental ventral → núcleo accumbens) están tónicamente inhibidas por interneuronas GABAérgicas. Las drogas de abuso suprimen esta inhibición, ya sea directamente (etanol, benzodiacepinas) o indirectamente (opioides: activan receptores µ en las interneuronas GABAérgicas, silenciándolas). El resultado es una liberación masiva de dopamina en el núcleo accumbens que activa los circuitos de recompensa del sistema límbico y produce la sensación de euforia.
El uso continuado de drogas produce tolerancia y dependencia física.:
- Tolerancia: se produce principalmente por mecanismos de desensibilización y reducción del número de receptores disponibles (internalización y down-regulation): se necesita más sustancia para activar los receptores que quedan.
- Dependencia física: se manifiesta al suprimir bruscamente la droga. El SNC, que ha reducido su sensibilidad para compensar el efecto de la sustancia, queda transitoriamente sin la inhibición GABAérgica (o dopaminérgica) que necesita, produciendo síntomas de abstinencia inversos al efecto de la droga (agitación, convulsiones, taquicardia en el caso del alcohol; ansiedad, insomnio en el caso de las benzodiazepinas).
Un error frecuente es describir la tolerancia como «síntesis aumentada de receptores». Es exactamente lo contrario: la tolerancia a los opioides, las benzodiazepinas y el alcohol se produce por down-regulation (reducción del número de receptores disponibles), lo que obliga a aumentar la dosis para obtener el mismo efecto.
El sistema límbico contiene una alta densidad de neuronas GABAérgicas. Las drogas que actúan sobre estas neuronas producen efectos sobre el estado emocional y pueden precipitar episodios psicóticos en sujetos vulnerables.
