24.1. Diferenciación de la gónada indiferenciada
24.1.1. Cordones sexuales primarios y secundarios
Hasta la sexta semana de desarrollo, la gónada es una estructura bipotencial capaz de convertirse en testículo o en ovario según la señal molecular que reciba.
Del epitelio celómico surgen inicialmente unos cordones sexuales primarios que rodean a las células germinales primordiales, ya llegadas a la gónada tras su migración desde el epiblasto.
Los cordones primarios desaparecen y son sustituidos por cordones sexuales secundarios, organizados en dos poblaciones con topografía opuesta: una medular (central) y otra cortical (periférica).
24.1.2. La vía SRY-SOX9 y la ruta ovárica por defecto
El gen SRY, localizado en el brazo corto del cromosoma Y (Yp11.3), codifica un factor de transcripción que activa la expresión de SOX9 en las células de la cresta gonadal. SOX9 dirige la diferenciación de esas células hacia células de Sertoli, que a su vez organizan los cordones medulares en los futuros túbulos seminíferos. En presencia de esta vía, los cordones medulares persisten y los corticales se atrofian: se forma un testículo.
En ausencia de SRY, la gónada sigue su ruta por defecto. Persisten los cordones corticales y se atrofian los medulares, con los folículos primarios manteniéndose en la periferia hasta la pubertad, momento en el que las hormonas iniciarán la ovulación y la menstruación.
Un error conceptual habitual es imaginar la determinación sexual como dos programas genéticos igualmente activos que compiten entre sí.
En realidad, la vía SRY-SOX9 es la única señal activa necesaria; sin ella, el desarrollo ovárico ocurre por defecto. Esto explica por qué mutaciones que inactivan SRY en un individuo XY producen fenotipo femenino (disgenesia gonadal XY, síndrome de Swyer), mientras que la translocación de SRY a un cromosoma X produce fenotipo masculino en un individuo XX.
Los cordones medulares del testículo, en su porción más próxima al hilio, dan lugar a la red de Haller (rete testis), la red de conductillos que conectará posteriormente con las vías excretoras derivadas del conducto de Wolf.
24.2. Aparato genital masculino
24.2.1. Derivados del conducto de Wolf
Las vías excretoras masculinas se forman a partir del conducto mesonéfrico o conducto de Wolf, presente desde el desarrollo del mesonefros (ver tema 23). Bajo la influencia de la testosterona secretada por las células de Leydig fetales, el conducto de Wolf se diferencia a cada lado en epidídimo, conducto deferente, vesícula seminal y conducto eyaculador, entre la octava y la decimotercera semana.
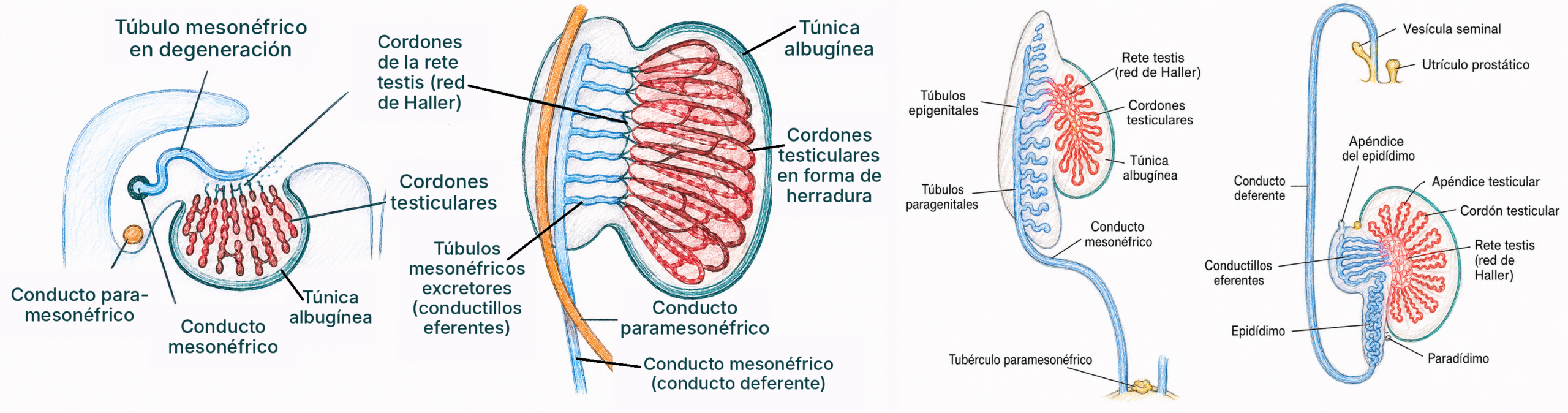
Cuando el conducto de Wolf se diferencia con normalidad en epidídimo y vesícula seminal pero falla específicamente en el segmento del conducto deferente, el resultado es la ausencia congénita bilateral de deferentes (ACBD), presente en más del 95% de los varones con fibrosis quística por mutación de CFTR.
No afecta a la producción de espermatozoides en el testículo, solo a su vía de salida: es causa de azoospermia obstructiva, no secretora, y es tratable mediante extracción testicular de espermatozoides para técnicas de reproducción asistida.
24.2.2. Regresión mülleriana
La AMH (también llamada sustancia inhibidora mülleriana (MIS)) es una glicoproteína secretada por las células de Sertoli desde la octava semana de desarrollo.
Su función es provocar la regresión del conducto de Müller en el varón, un proceso independiente de la acción de la testosterona sobre el conducto de Wolf.
Son dos hormonas distintas actuando sobre dos conductos distintos, en paralelo.
El conducto de Müller, pese a esta regresión, no desaparece de forma absoluta, sino que persiste de forma vestigial en dos estructuras:
- El utrículo prostático.
- Las hidátides de Morgagni.
Son restos sin función, pero ocasionalmente son origen de quistes paratesticulares.
Cuando existe una mutación en el gen de la AMH o en su receptor, la regresión mülleriana no se produce pese a una diferenciación testicular y una virilización externa completamente normales.
El resultado es un varón genética y fenotípicamente normal que, sin embargo, conserva trompas de Falopio, útero y porción superior de vagina, habitualmente descubiertos de forma incidental durante una cirugía por criptorquidia o hernia inguinal.
24.3. Aparato genital femenino
24.3.1. Formación del cuerpo de Müller
En ausencia de AMH, el conducto de Wolf se elimina por falta de estímulo androgénico y los conductos de Müller persisten como estructura principal. Ambos conductos, con forma de dedo de guante, crecen en sentido caudal, se aproximan en la línea media y se fusionan formando una única estructura, el cuerpo de Müller, origen de:
- Las trompas de Falopio.
- El útero.
- El tercio superior de la vagina.
El conducto de Wolf, por su parte, no desaparece del todo en la mujer, puede persistir como:
- El paraoóforo, junto a las trompas.
- Los cuerpos de Gartner, junto al útero.
Son restos vestigiales equivalentes a los que el varón conserva del conducto de Müller.
Malformaciones de fusión mülleriana
Un fallo de fusión entre las dos mitades del cuerpo de Müller produce un espectro de malformaciones:
- Útero bicorne: fusión parcial, dos cuernos con cuerpo y cérvix común.
- Útero bidelfo: fusión ausente, duplicación completa de cuerpo, cérvix y en ocasiones vagina.
- Útero septado: fusión completa pero reabsorción incompleta del tabique medial.
En conjunto, estas malformaciones afectan a un 4-7% de las mujeres y se asocian a mayor tasa de aborto espontáneo, parto pretérmino y presentaciones fetales anómalas.
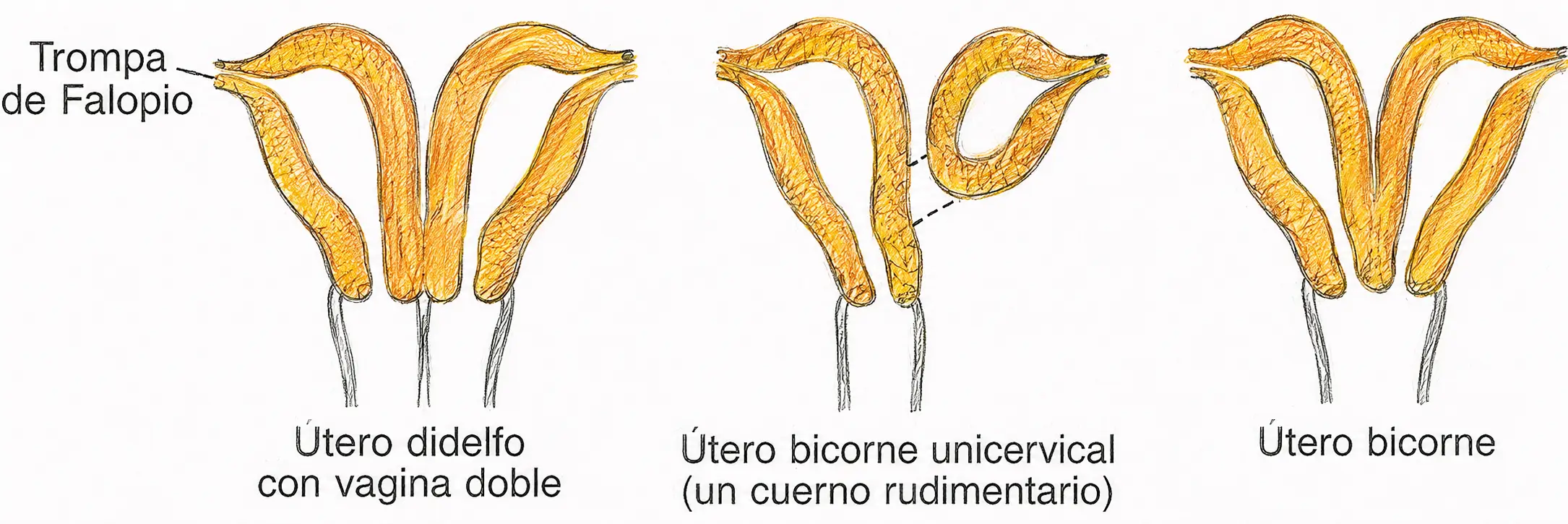
El fallo de fusión (bicorne, bidelfo, septado) implica que ambos conductos de Müller se formaron con normalidad pero no se unieron correctamente.
Es un mecanismo distinto de la agenesia mülleriana completa, como ocurre en el síndrome de Mayer-Rokitansky-Küster-Hauser (1 de cada 4.500 nacidas vivas), donde los conductos de Müller ni siquiera llegan a desarrollarse, con ausencia de útero y vagina superior en una mujer con ovarios y cariotipo XX normales.
24.3.2. Formación de la vagina y el himen
La vagina tiene un origen mixto:
- Su tercio superior es mülleriano.
- Sus dos tercios inferiores derivan del seno urogenital.
Inicialmente es un cordón macizo de posición horizontal que progresivamente se verticaliza y se une al ectodermo externo, formando en ese punto de contacto el himen.
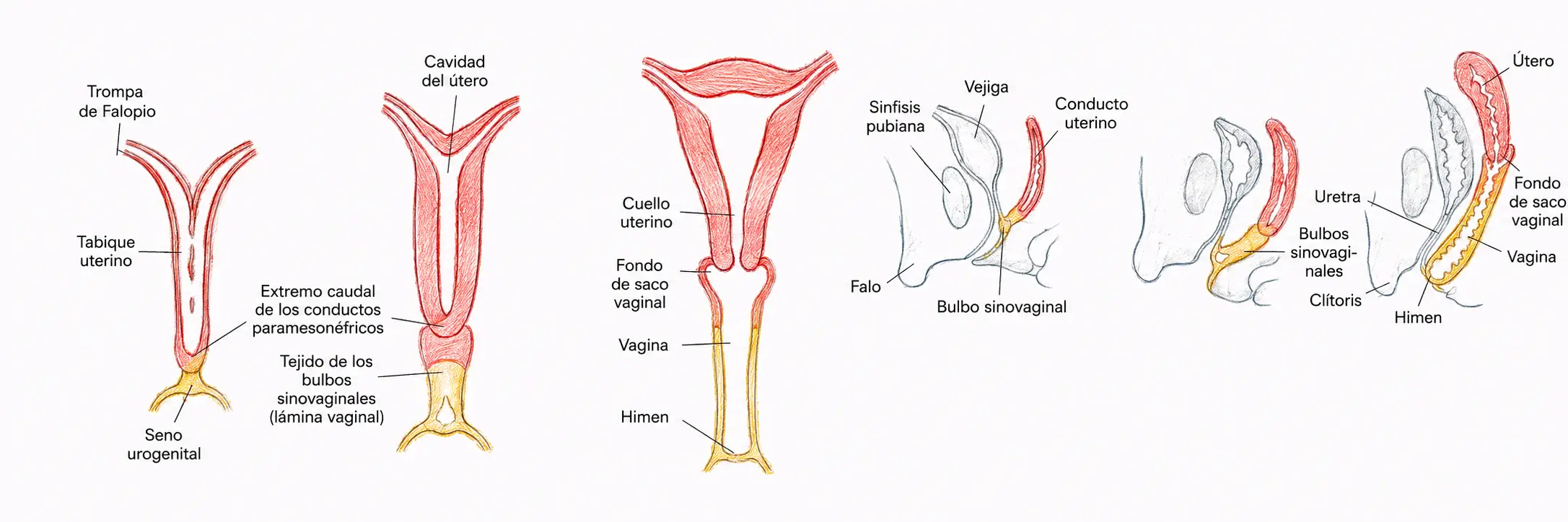
El himen es la zona de contacto entre la vagina (de origen mesodérmico-mülleriano y sinusal) y el ectodermo externo, no una estructura formada exclusivamente por uno de los dos tejidos. Su cierre completo, sin perforación (himen imperforado), retiene el flujo menstrual y se presenta clínicamente como amenorrea primaria con dolor pélvico cíclico.
24.4. Descenso gonadal
24.4.1. El gubernáculo genital en ambos sexos
Ambos sexos comparten una misma estructura directora de la migración gonadal, el gubernáculo genital (un cordón mesenquimal que conecta el polo caudal de la gónada con la región inguinal desde etapas tempranas del desarrollo, mucho antes de que exista diferenciación sexual completa). Su destino final diverge:
- En la mujer, el gubernáculo se transforma en el ligamento útero-ovárico (fija el ovario al útero) y en el ligamento redondo del útero (lo une a los labios mayores).
- En el varón, el mismo gubernáculo arrastra al testículo, formado inicialmente a nivel lumbar, hasta su posición definitiva en el escroto.
En la mujer, sin el estímulo de INSL3 (insulin-like factor 3) que sí actúa en el varón (→ apartado 24.4.2), el gubernáculo se transforma en el ligamento útero-ovárico, que fija el ovario al útero, y en el ligamento redondo del útero, que lo prolonga hasta los labios mayores.
En el varón, el mismo cordón, bajo la acción de INSL3, arrastra al testículo, formado inicialmente a nivel lumbar, hasta su posición definitiva en el escroto.
24.4.2. Descenso testicular en dos fases y criptorquidia
El testículo, durante su descenso, arrastra consigo capas de mesodermo que lo rodearán de forma permanente; entre ellas se forma la capa albugínea, la cápsula fibrosa que lo separa del epitelio externo. La bolsa escrotal, a diferencia de su contenido, procede del ectodermo.
El descenso testicular tiene una fase transabdominal (semanas 8-15), controlada principalmente por INSL3 (insulin-like factor 3) secretado por las células de Leydig, y una fase inguinoescrotal (semanas 25-35), dependiente de andrógenos. Esta segunda fase es la que atraviesa el canal inguinal hasta alcanzar el escroto definitivo.
La valoración real del descenso completo se hace tras la semana 35, al finalizar la fase inguinoescrotal. La criptorquidia afecta al 3-4% de los recién nacidos a término, cifra que sube hasta el 30% en prematuros (coherente con que la fase inguinoescrotal ocurre precisamente en el tercer trimestre).
No es un fallo de diferenciación testicular, sino exclusivamente de migración. Su persistencia tras el primer año de vida multiplica entre 2 y 8 veces el riesgo de cáncer testicular en la vida adulta y compromete significativamente la fertilidad futura.
24.5. Polo caudal y genitales externos
24.5.1. Estructuras indiferenciadas comunes
En el polo caudal externo, el mesodermo constriñe el tubo digestivo mediante los espolones mesodérmicos, dando lugar a tres estructuras comunes a ambos sexos antes de su diferenciación:
- Dos abultamientos laterales, los rodetes genitales.
- Un abultamiento medio impar, el tubérculo genital, rodeado por los pliegues uretrales.
El crecimiento del tubérculo genital depende de una señal de SHH (sonic hedgehog) procedente del endodermo cloacal, un mecanismo de señalización análogo al desarrollo del eje anteroposterior de la extremidad (ver tema 25, apartado 25.3.2). No es la misma estructura ni el mismo eje, pero es el mismo principio de un centro señalizador puntual dirigiendo el crecimiento de una protrusión mesenquimal.
24.5.2. Diferenciación masculina y femenina
En el varón, los rodetes genitales forman las bolsas testiculares, el tubérculo genital da origen al pene, y los pliegues uretrales se fusionan formando el cuerpo del pene y cerrando la uretra peneana.
En la mujer, sin el estímulo androgénico que impulsa esa fusión, los rodetes genitales forman los labios mayores, el tubérculo genital forma el clítoris, y los pliegues uretrales permanecen sin fusionar, dando lugar a los labios menores.
Cuando la fusión de los pliegues uretrales en el varón es incompleta, el meato uretral se abre en la cara ventral del pene en lugar de en su extremo distal. Esta malformación se denomina hipospadias.
Está presente en aproximadamente 1 de cada 200-300 nacidos varones, la malformación genital externa más frecuente en la especie humana.
| Estructura embrionaria | Derivado masculino | Derivado femenino (vestigio) | Patología asociada |
|---|---|---|---|
| Conducto de Wolf | Epidídimo, conducto deferente, vesícula seminal, conducto eyaculador | Paraoóforo, cuerpos de Gartner | Quistes de Gartner (femenino); ausencia congénita de deferentes en fibrosis quística (masculino) |
| Conducto de Müller | Utrículo prostático, hidátide de Morgagni | Trompas de Falopio, útero, 1/3 superior de vagina | Síndrome de persistencia de conductos müllerianos (masculino); útero bicorne/bidelfo, síndrome de Mayer-Rokitansky-Küster-Hauser (femenino) |
| Gubernáculo genital | Guía el descenso testicular al escroto | Ligamento útero-ovárico, ligamento redondo del útero | Criptorquidia (masculino) |
| Tubérculo genital / pliegues uretrales | Pene, cuerpo peneano por fusión de pliegues | Clítoris, labios menores (pliegues sin fusionar) | Hipospadias (masculino, 1/200-300) |
